Publications
publications by categories in reversed chronological order. generated by jekyll-scholar.
2026
-
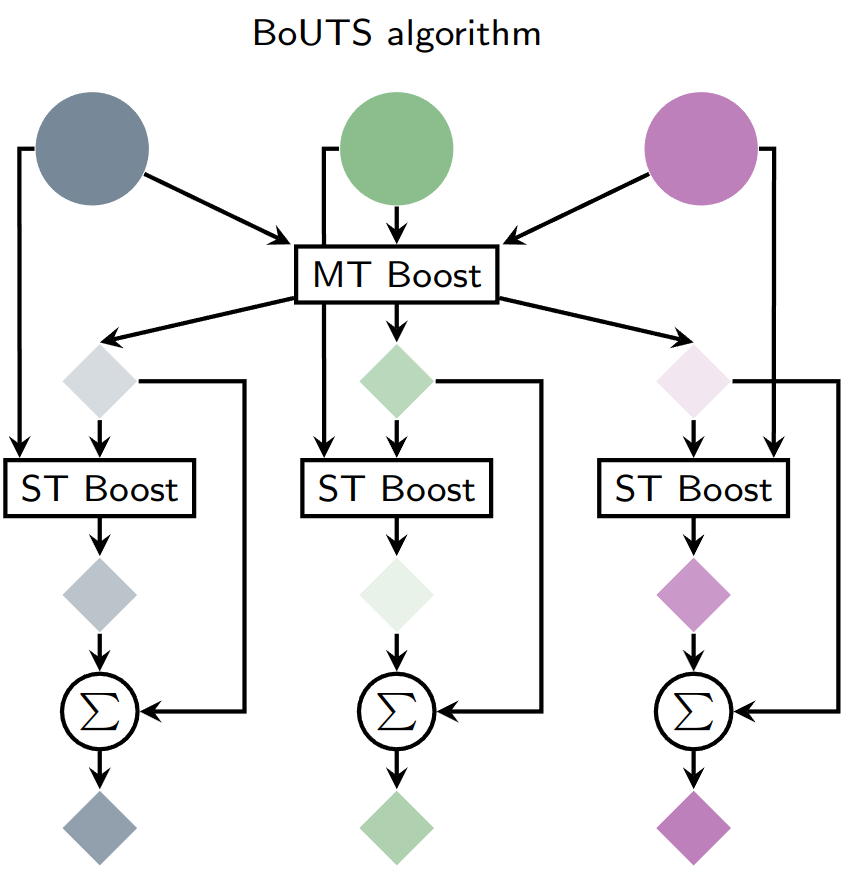 Universal Feature Selection for Simultaneous Interpretability of Multitask DatasetsMatt Raymond, Jacob Charles Saldinger, Paolo Elvati, and Angela Violi2026
Universal Feature Selection for Simultaneous Interpretability of Multitask DatasetsMatt Raymond, Jacob Charles Saldinger, Paolo Elvati, and Angela Violi2026Extracting meaningful features from complex, high-dimensional datasets across scientific domains remains challenging. Current methods often struggle with scalability, limiting their applicability to large datasets, or make restrictive assumptions about feature-property relationships, hindering their ability to capture complex interactions. BoUTS’s general and scalable feature selection algorithm surpasses these limitations to identify both universal features relevant to all datasets and task-specific features predictive for specific subsets. Evaluated on seven diverse chemical regression datasets, BoUTS achieves state-of-the-art feature sparsity while maintaining prediction accuracy comparable to specialized methods. Notably, BoUTS’s universal features enable domain-specific knowledge transfer between datasets, and suggest deep connections in seemingly-disparate chemical datasets. We expect these results to have important repercussions in manually-guided inverse problems. Beyond its current application, BoUTS holds immense potential for elucidating data-poor systems by leveraging information from similar data-rich systems. BoUTS represents a significant leap in cross-domain feature selection, potentially leading to advancements in various scientific fields.
2025
-
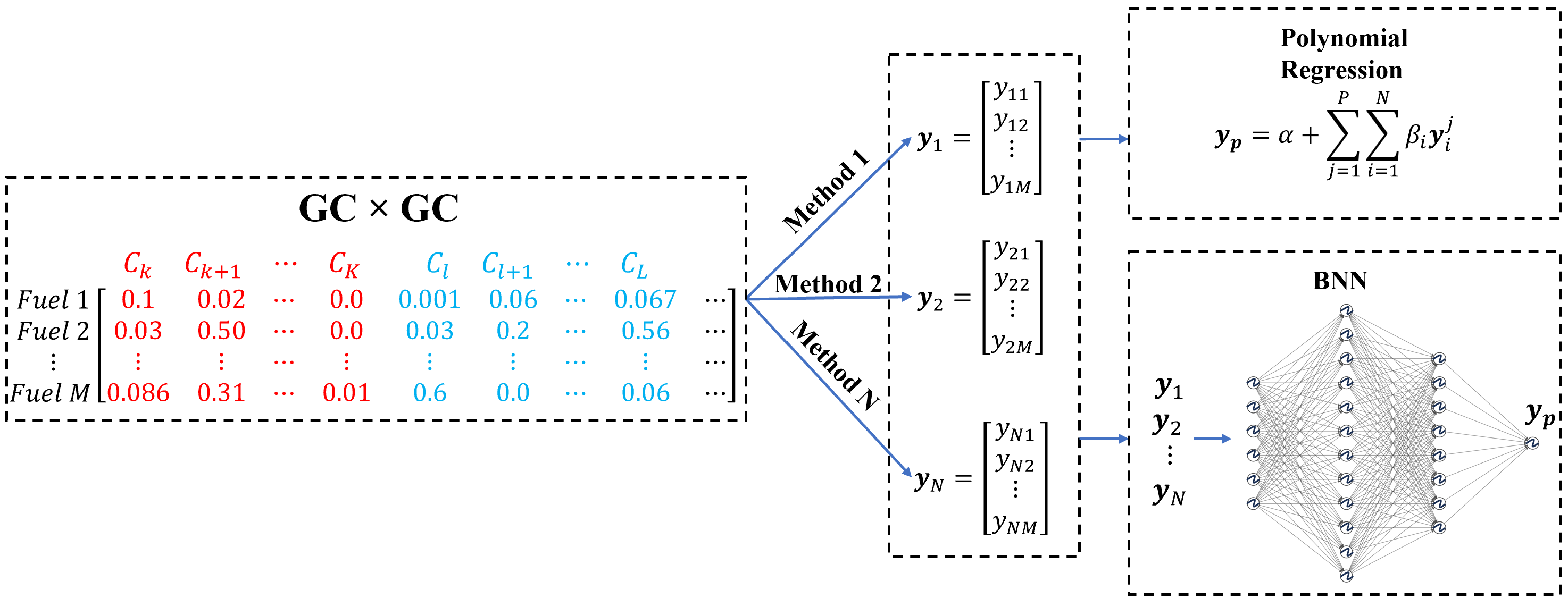 A Bayesian Ensemble Approach for Improved Sustainable Aviation Fuel ModelingMohammed I. Radaideh, Majdi I. Radaideh, and A. VioliEnergy Conversion and Management: X, 2025Revised Manuscript Submitted, Manuscript Number: ECMX-D-25-00548
A Bayesian Ensemble Approach for Improved Sustainable Aviation Fuel ModelingMohammed I. Radaideh, Majdi I. Radaideh, and A. VioliEnergy Conversion and Management: X, 2025Revised Manuscript Submitted, Manuscript Number: ECMX-D-25-00548In this work, we introduce a new methodology to combine the available methods to predict the properties of complex hydrocarbon mixtures such as aviation fuels. Due to the complexity of aviation fuels, the available methods perform well individually on some of the experimental observations and vice versa on others when a surrogate aviation fuel is defined and used. To this end, we introduce a new ensemble model based on the existing methods that combine and weigh their predictions. We employ the probabilistic Bayesian approach to predict aviation fuel properties with confidence levels. This is necessary because the available experimental data for aviation fuels is generally limited, which leads to overfitting. We adopt both “interpretable” Bayesian regression and a more “black-box” approach to Bayesian neural networks. An ensemble of predictive methods provided better predictions than the individual methods with robust confidence levels for three properties considered: mass density, kinematic viscosity, and flash point. A significant reduction in the mean absolute percentage error was obtained for mass density predictions, from 1.25% to 0.57% and 0.42%, using the Bayesian linear regression (BLR) and Bayesian Neural Network (BNN), respectively. The error in kinematic viscosity predictions was reduced from 17.25% to 9.02% and 6.79% using BLR and BNN, respectively. The error in flash point predictions is reduced from 9.04% to 5.83% by BLR and to 5.51% by BNN. The importance of the methods in the ensemble did not fully follow their individual performance, where the accurate models may not be the most important. The ensemble approach allows for the inclusion of new methods, even if they are slightly less accurate. This methodology can be extended to predict other aviation fuel properties and incorporate any predictive model. It also offers a way to generate valid training data for generative Artificial Intelligence (AI) models, helping to address the scarcity of aviation fuel data.
-
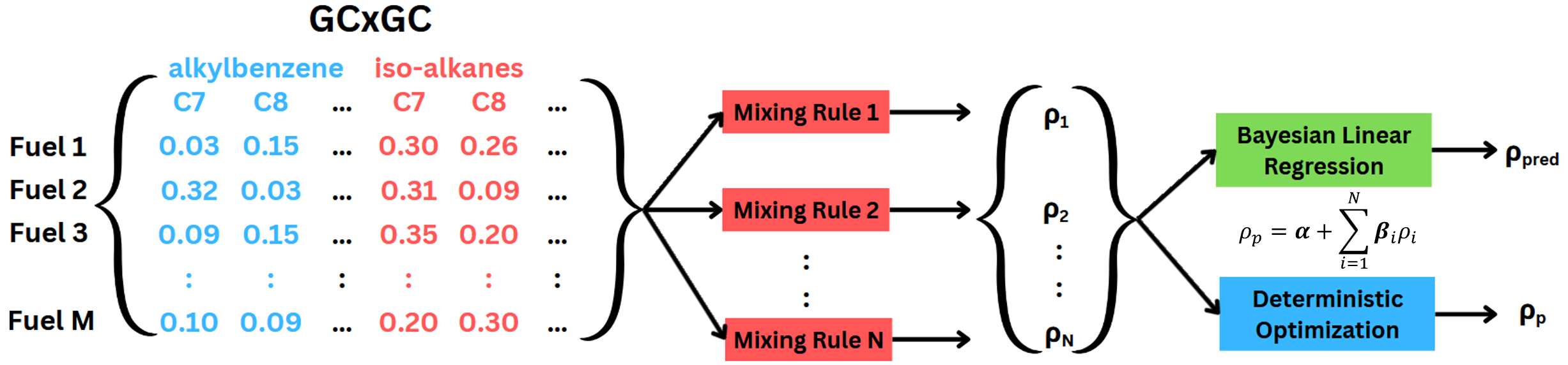 A Bayesian Approach to Predict Sustainable Aviation Fuels Properties Using an Ensemble of Property MethodsMohammed I. Radaideh, Majdi I. Radaideh, and A. VioliIn AIAA AVIATION Forum and Exposition, Jul 2025
A Bayesian Approach to Predict Sustainable Aviation Fuels Properties Using an Ensemble of Property MethodsMohammed I. Radaideh, Majdi I. Radaideh, and A. VioliIn AIAA AVIATION Forum and Exposition, Jul 2025We propose in this work a new methodology for predicting aviation fuel properties by combining the predictions of existing methods into an ensemble. The performance of current methods varies among different types of aviation fuels, including conventional jet fuels, Sustainable Aviation Fuels (SAF), and biofuels. Aviation fuels are a complex mixture of several hydrocarbons, which may also lead to high prediction errors. The predictions of the methods in the ensemble are weighted, where the weights are obtained using a probabilistic Bayesian linear regression (BLR) model. We tested the methodology used to predict the mass density of different aviation fuels. We also compared the results of BLR with those of deterministic optimization using Particle Swarm Optimization (PSO). Both BLR and PSO reduced the prediction error of mass density from 1.24% to 0.57% and 0.49%, respectively. We noticed a trade-off between uncertainty and accuracy in BLR results, where higher accuracy is accompanied by higher uncertainty. Accordingly, more accurate results provided by PSO or possibly other deterministic approaches may be misleading, and uncertainty should be considered to draw more robust conclusions. Results also showed that the individual performance of the methods does not follow their importance in the ensemble, i.e., the most accurate models do not necessarily have the highest weights; less accurate models could be more important. As a result, new predicting methods can be added even if they are, to some extent, less accurate than the methods in the ensemble.
-
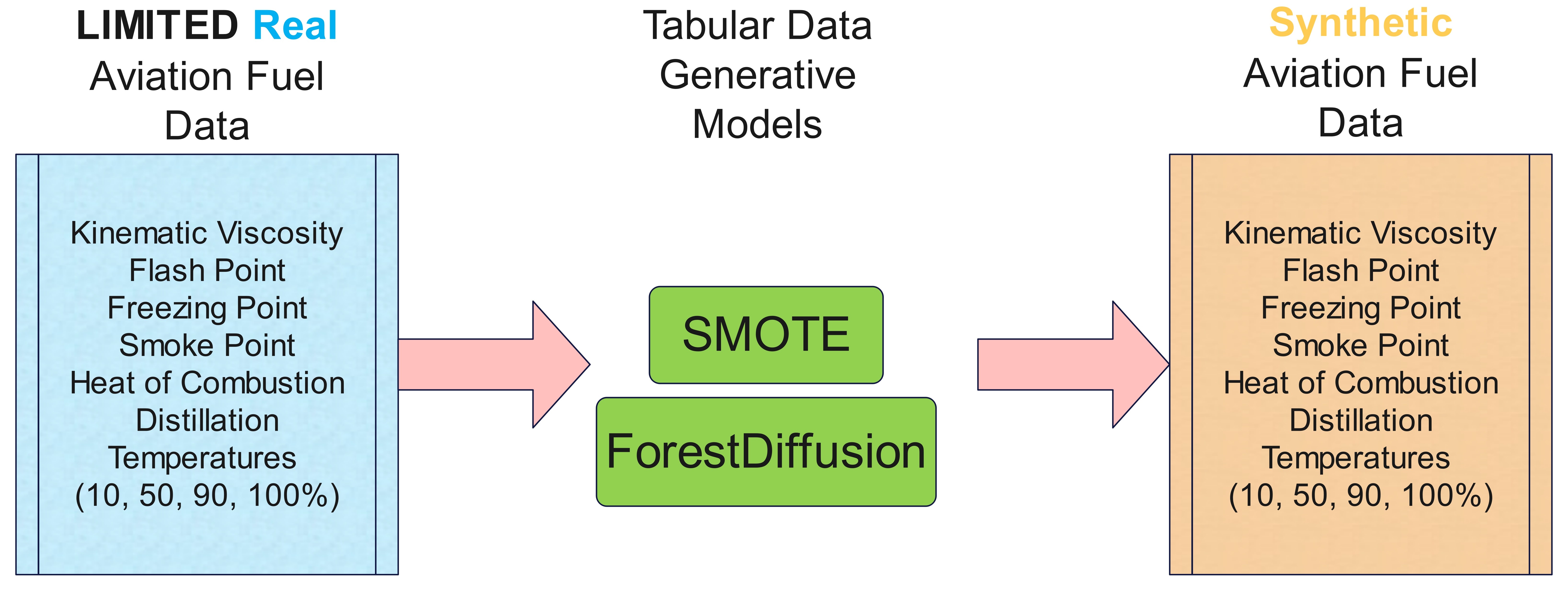 Generative AI Approach for Synthetic Aviation Fuels Data GenerationMohammed I. Radaideh, Majdi I. Radaideh, and A. VioliIn International Mechanical Engineering Congress & Exposition, Nov 2025
Generative AI Approach for Synthetic Aviation Fuels Data GenerationMohammed I. Radaideh, Majdi I. Radaideh, and A. VioliIn International Mechanical Engineering Congress & Exposition, Nov 2025Data scarcity is a common problem related to aviation fuels. Many studies are still developing predictive models to predict aviation fuel properties using fuel composition to help in Sustainable Aviation Fuels (SAF) certification. However, due to the data scarcity, most of these models were trained using limited experimental data and are prone to overfitting. To this end, we utilize generative AI models as a data augmentation technique to address the data scarcity of aviation fuels. The tabular data of aviation fuels considered includes experimental data for fuel composition and nine thermophysical properties. This data is used to train ForestDiffusion, a recently developed diffusion generative AI model for tabular data generation without the need for many training samples. The performance of ForestDiffusion is compared with another simple model, Synthetic Minority Oversampling TEchnique (SMOTE), that is commonly used as a baseline. Different metrics are used to assess the quality of the synthetic data generated by ForestDiffusion and SMOTE. Results showed that the simple model SMOTE provided better synthetic data fidelity, diversity, and machine learning utility. Still, it has lower real data privacy protection than ForestDiffusion, making the latter a more desirable choice regardless of the advantages that SMOTE provided, and showing the importance of considering multiple assessment metrics to have more balanced performance conclusions.
-
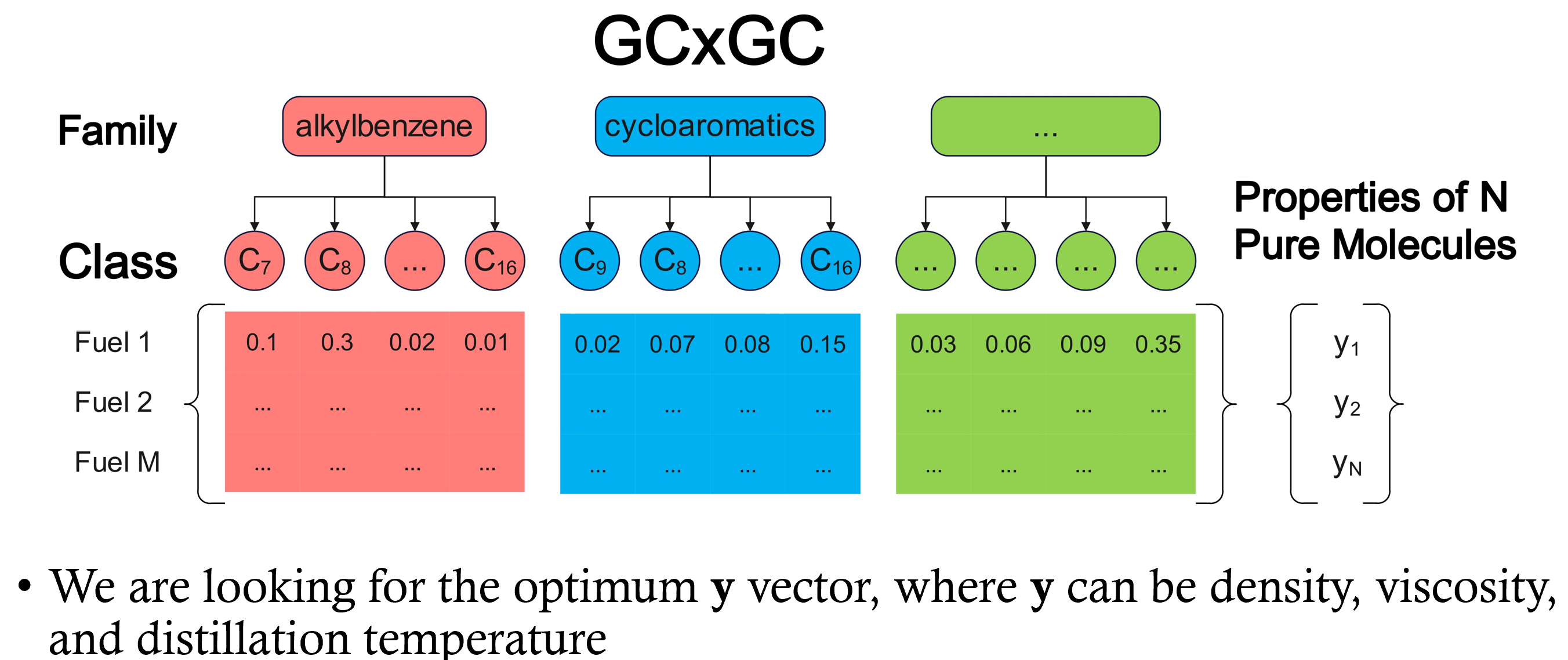 Optimal Sets of Molecules to Predict Aviation Fuel PropertiesMohammed I. Radaideh, D. Kim, Majdi I. Radaideh, and A. VioliIn 2025 Energy & Propulsion Conference & Exhibition, Oct 2025
Optimal Sets of Molecules to Predict Aviation Fuel PropertiesMohammed I. Radaideh, D. Kim, Majdi I. Radaideh, and A. VioliIn 2025 Energy & Propulsion Conference & Exhibition, Oct 2025The complexity and variability of modern aviation fuels necessitate the development of robust and efficient tools to assess their properties accurately, particularly within the certification framework established by the American Society for Testing and Materials (ASTM). Therefore, previous research has developed predictive models to reduce the experimental burden by predicting aviation fuel properties from broad chemical classes. While two-dimensional Gas Chromatography (GC\timesGC) provides detailed compositional information, it only identifies the weight of hydrocarbon families (aromatics, cycloalkanes, n-alkanes, iso-alkanes), not individual molecules. Aviation fuels are complex, and their composition can contain more than 60 key classes. As a result, an exceptionally high number of possible molecule combinations makes random selection prone to high errors in property prediction. We will use a Monte Carlo approach to search for the optimal combination of 64 hydrocarbon molecules from this vast combinatorial space. By exploring up to 500 million combinations, we aim to determine the molecule set that best predicts mass density, kinematic viscosity, and heat of combustion using linear mixing rules. These rules calculate the properties of molecule mixtures using the weight of each molecule in the mixture and the pure molecules’ properties. We will utilize experimental data for various aviation fuels, including conventional jet fuels, sustainable aviation fuels, and rocket propulsion fuels. This research benefits the surrogate fuel analysis and will provide insights into the best isomers or molecules to predict aviation fuel properties with the least error. This work will help deliver aviation fuel producers with a relatively accurate pre-screening tool for property prediction, minimizing the need for iterative experimental processes.
-
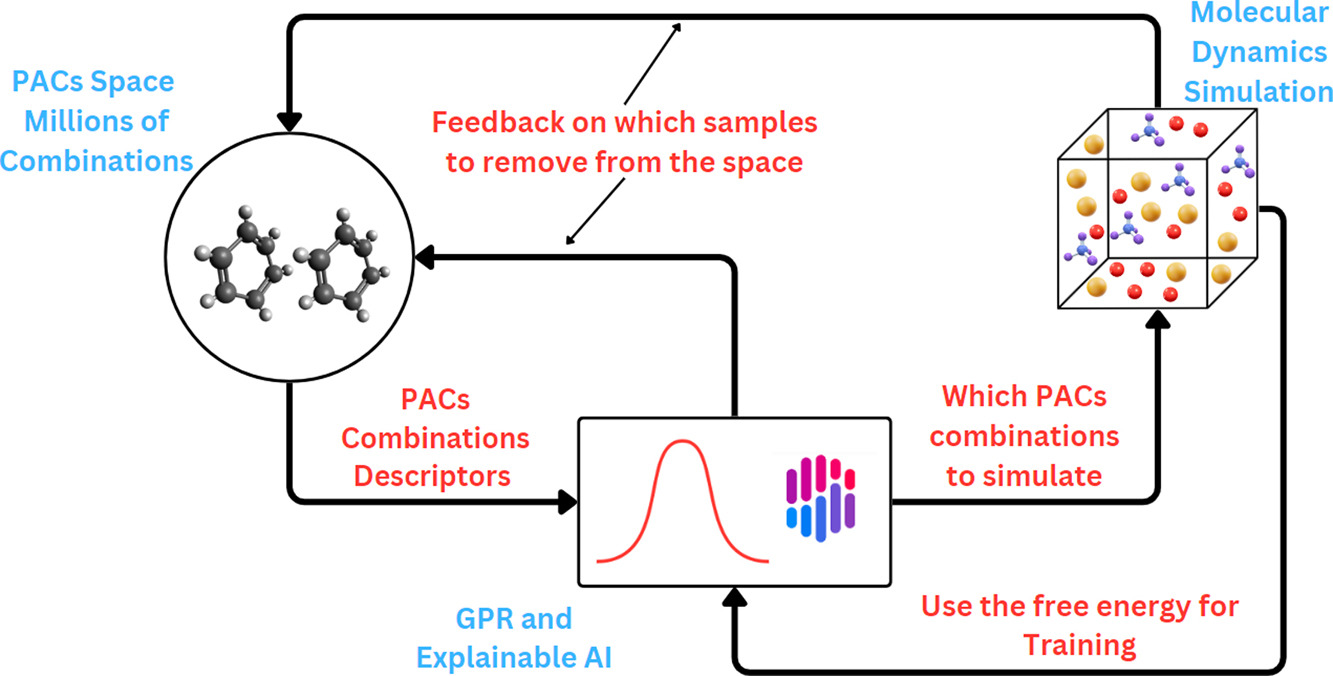 Efficient sampling of polycyclic aromatic compounds for free energy predictions through active learningMohammed I. Radaideh, Matt Raymond, Paolo Elvati, Jacob C Saldinger, Majdi I. Radaideh, and 1 more authorEnergy and AI, Oct 2025
Efficient sampling of polycyclic aromatic compounds for free energy predictions through active learningMohammed I. Radaideh, Matt Raymond, Paolo Elvati, Jacob C Saldinger, Majdi I. Radaideh, and 1 more authorEnergy and AI, Oct 2025The physical growth of Polycyclic Aromatic Compounds (PACs) to soot particles plays a significant role in understanding the chemistry of soot formation. Insights into the process can be gained from PACs’ free energy of dimerization landscape. However, because the infeasibly large space of possible PAC dimers cannot be exhaustively simulated, researchers must train machine learning models on a subset of data to impute the rest. To this end, we propose and assess an active learning approach to discovering the optimal PACs for training a machine learning model to predict PACs’ association and dissociation free energies. The comparison between active learning and random sampling showed that active learning has faster loss convergence, requiring fewer training samples to reach the same level of accuracy. The trained model accurately modeled unseen PACs and exhibited robustness against changes in the sampling space used to train the model. More broadly, this work shows how active learning can optimize the design and improve the understanding of more expensive models in specific domains.
- Joint Diffusion Sampling via Positive-Unlabeled Guidance for Multi-Modal DataMatt Raymond, Yilun Zhu, Jianxin Zhang, Angela Violi, and Clayton ScottIn Proceedings of the ICML 2025 Workshop on Foundation Models for Life Sciences (FM4LS), Jul 2025
Multi-modal generative models typically require abundant training data from multi-modal joint distributions, which is often unavailable in the life sciences. We propose to treat each modality as a marginal distribution and correct their independent diffusion processes to sample from their joint distribution. Specifically, we introduce "joint diffusion sampling," a method that generates a sample from joint distributions using pre-trained models for individual (uni-modal) marginal distributions and minimal data from the (multi-modal) joint distribution. We demonstrate preliminary uni- and multi-modal results for images, molecules, and Boolean values, and discuss multi-modal applications of our approach.
-
 Machine learning models for Si nanoparticle growth in nonthermal plasmaMatt Raymond, Paolo Elvati, Jacob Charles Saldinger, Jonathan Lin, Xuetao Shi, and 1 more authorPlasma Sources Science and Technology, Jul 2025
Machine learning models for Si nanoparticle growth in nonthermal plasmaMatt Raymond, Paolo Elvati, Jacob Charles Saldinger, Jonathan Lin, Xuetao Shi, and 1 more authorPlasma Sources Science and Technology, Jul 2025Nanoparticles (NPs) formed in nonthermal plasmas (NTPs) can have unique properties and applications. However, modeling their growth in these environments presents significant challenges due to the non-equilibrium nature of NTPs, making them computationally expensive to describe. In this work, we address the challenges associated with accelerating the estimation of parameters needed for these models. Specifically, we explore how different machine learning models can be tailored to improve prediction outcomes. We apply these methods to reactive classical molecular dynamics data, which capture the processes associated with colliding silane fragments in NTPs. These reactions exemplify processes where qualitative trends are clear, but their quantification is challenging, hard to generalize, and requires time-consuming simulations. Our results demonstrate that good prediction performance can be achieved when appropriate loss functions are implemented and correct invariances are imposed. While the diversity of molecules used in the training set is critical for accurate prediction, our findings indicate that only a fraction (15-25%) of the energy and temperature sampling is required to achieve high levels of accuracy. This suggests a substantial reduction in computational effort is possible for similar systems.
2024
- SPIN: A Data-Driven Model to Reduce Large Chemical Reaction NetworksMayank Baranwal, Jacob C. Saldinger, Doohyun Kim, Paolo Elvati, Alfred O. Hero, and 1 more authorFuel, Jul 2024
-
 Can Machine Learning Overcome the 95% Failure Rate and Reality that Only 30% of Approved Cancer Drugs Meaningfully Extend Patient Survival?Duxin Sun, Christian Macedonia, Zhigang Chen, Sriram Chandrasekaran, Kayvan Najarian, and 12 more authorsJournal of Medicinal Chemistry, Sep 2024
Can Machine Learning Overcome the 95% Failure Rate and Reality that Only 30% of Approved Cancer Drugs Meaningfully Extend Patient Survival?Duxin Sun, Christian Macedonia, Zhigang Chen, Sriram Chandrasekaran, Kayvan Najarian, and 12 more authorsJournal of Medicinal Chemistry, Sep 2024Despite implementing hundreds of strategies, cancer drug development suffers from a 95% failure rate over 30 years, with only 30% of approved cancer drugs extending patient survival beyond 2.5 months. Adding more criteria without eliminating nonessential ones is impractical and may fall into the “survivorship bias” trap. Machine learning (ML) models may enhance efficiency by saving time and cost. Yet, they may not improve success rate without identifying the root causes of failure. We propose a “STAR-guided ML system” (structure-tissue/cell selectivity-activity relationship) to enhance success rate and efficiency by addressing three overlooked interdependent factors: potency/specificity to the on/off-targets determining efficacy in tumors at clinical doses, on/off-target-driven tissue/cell selectivity influencing adverse effects in the normal organs at clinical doses, and optimal clinical doses balancing efficacy/safety as determined by potency/specificity and tissue/cell selectivity. STAR-guided ML models can directly predict clinical dose/efficacy/safety from five features to design/select the best drugs, enhancing success and efficiency of cancer drug development.
-
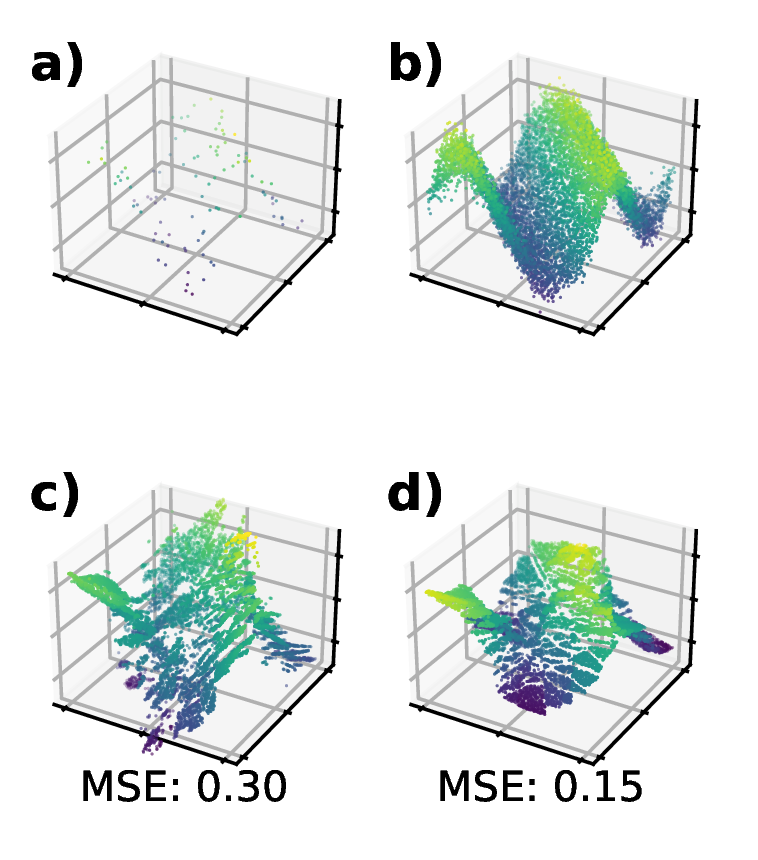 Joint Optimization of Piecewise Linear EnsemblesMatt Raymond, Angela Violi, and Clayton ScottIn IEEE 34th International Workshop on Machine Learning for Signal Processing, Sep 2024
Joint Optimization of Piecewise Linear EnsemblesMatt Raymond, Angela Violi, and Clayton ScottIn IEEE 34th International Workshop on Machine Learning for Signal Processing, Sep 2024Tree ensembles achieve state-of-the-art performance despite being greedily optimized. Global refinement (GR) reduces greediness by jointly and globally optimizing all constant leaves. We propose Joint Optimization of Piecewise Linear ENsembles (JOPLEN), a piecewise-linear extension of GR. Compared to GR, JOPLEN improves model flexibility and can apply common penalties, including sparsity-promoting matrix norms and subspace-norms, to nonlinear prediction. We evaluate the Frobenius norm, ℓ2,1 norm, and Laplacian regularization for 146 regression and classification datasets; JOPLEN, combined with GB trees and RF, achieves superior performance in both settings. Additionally, JOPLEN with a nuclear norm penalty empirically learns smooth and subspace-aligned functions. Finally, we perform multitask feature selection by extending the Dirty LASSO. JOPLEN Dirty LASSO achieves a superior feature sparsity/performance tradeoff to linear and gradient boosted approaches. We anticipate that JOPLEN will improve regression, classification, and feature selection across many fields.
-
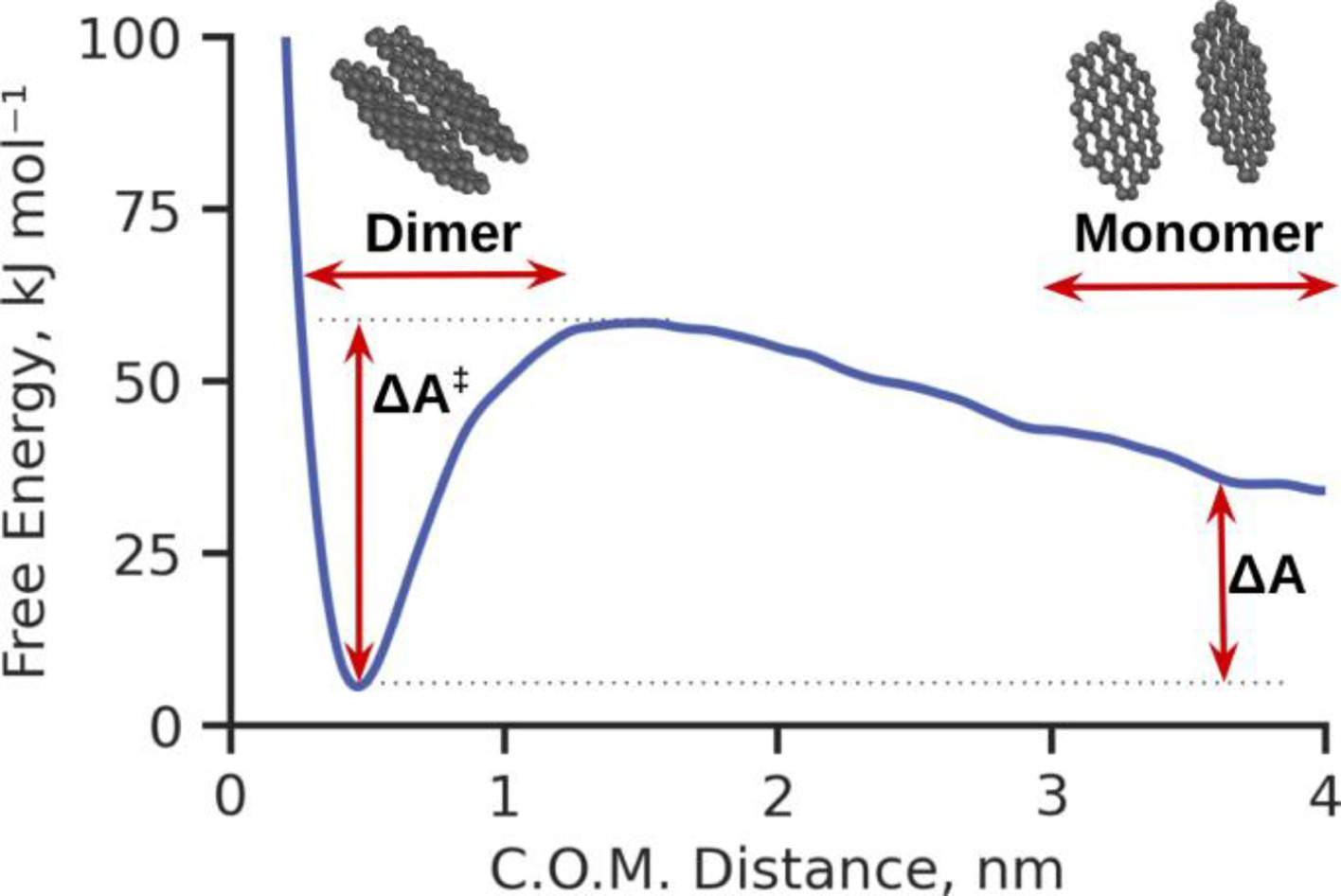 Predicting aggregation rates of polycyclic aromatics through machine learningJacob C. Saldinger, Paolo Elvati, Karam Alrawi, and Angela VioliFuel, Sep 2024
Predicting aggregation rates of polycyclic aromatics through machine learningJacob C. Saldinger, Paolo Elvati, Karam Alrawi, and Angela VioliFuel, Sep 2024The transition of gas-phase polycyclic aromatic compounds (PACs) into mature soot particles stands as a critical stage in the process of soot formation. However, formulating an accurate estimation of the rates of these processes poses a challenge, stemming from the need to consider the energy barriers controlling the dimerization of PACs. In this study, we underscore the effectiveness of machine learning in predicting the free energy barrier of dimer dissociation, which is fundamental to the dimerization rate, relying solely on the basic molecular features of any PAC. To obtain the free energy surfaces of the dimerization process for a diverse set of PACs at various temperatures, we employed well-tempered Metadynamics simulations accompanied by a gauge correction. Following this, we trained a machine learning model to predict free energy barriers across the said temperatures. We found that this model significantly outperforms the accuracy of commonly used mass correlations. Our machine-learning model also automatically identifies specific molecular descriptors. These descriptors quantify the relation to the barrier and qualitatively link to interpretable properties vital to the dimerization process. The study has far-reaching implications for soot formation research. It introduces an accurate method for estimating PAC dimerization rates, offering potential enhancements in soot formation model precision. Moreover, it pinpoints key molecular characteristics that provide a deeper comprehension of the mechanisms governing PAC clustering. This understanding can fuel the development of novel strategies for controlling soot formation.
2023
-
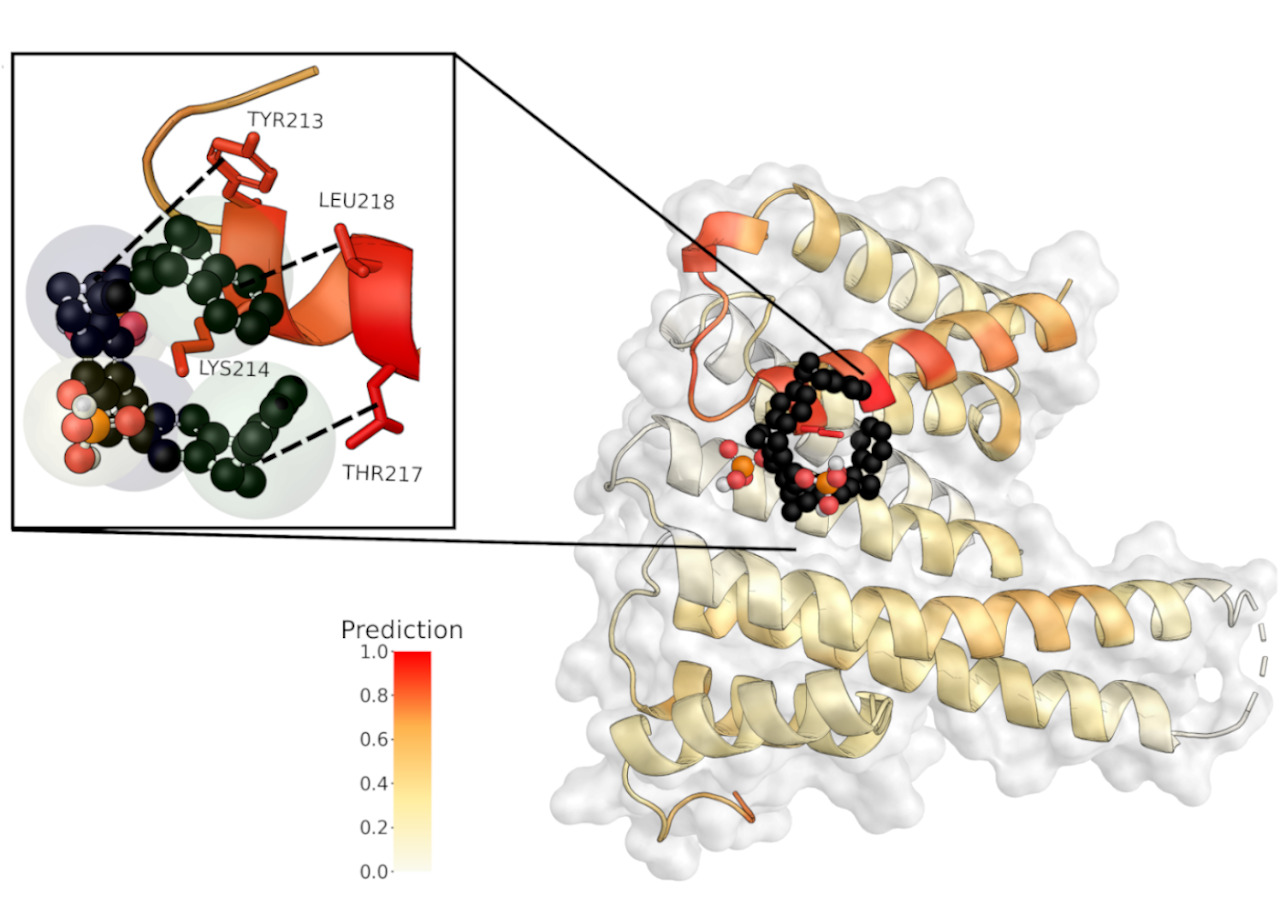 Domain-agnostic predictions of nanoscale interactions in proteins and nanoparticlesJacob Charles Saldinger, Matt Raymond, Paolo Elvati, and Angela VioliNature Computational Science, May 2023
Domain-agnostic predictions of nanoscale interactions in proteins and nanoparticlesJacob Charles Saldinger, Matt Raymond, Paolo Elvati, and Angela VioliNature Computational Science, May 2023Although challenging, the accurate and rapid prediction of nanoscale interactions has broad applications for numerous biological processes and material properties. While several models have been developed to predict the interaction of specific biological components, they use system-specific information that hinders their application to more general materials. Here we present NeCLAS, a general and efficient machine learning pipeline that predicts the location of nanoscale interactions, providing human-intelligible predictions. NeCLAS outperforms current nanoscale prediction models for generic nanoparticles up to 10–20\thinspacenm, reproducing interactions for biological and non-biological systems. Two aspects contribute to these results: a low-dimensional representation of nanoparticles and molecules (to reduce the effect of data uncertainty), and environmental features (to encode the physicochemical neighborhood at multiple scales). This framework has several applications, from basic research to rapid prototyping and design in nanobiotechnology.
-
 Low-THz Vibrations of Biological MembranesChloe Luyet, Paolo Elvati, Jordan Vinh, and Angela VioliMembranes, Jan 2023
Low-THz Vibrations of Biological MembranesChloe Luyet, Paolo Elvati, Jordan Vinh, and Angela VioliMembranes, Jan 2023A growing body of work has linked key biological activities to the mechanical properties of cellular membranes, and as a means of identification. Here, we present a computational approach to simulate and compare the vibrational spectra in the low-THz region for mammalian and bacterial membranes, investigating the effect of membrane asymmetry and composition, as well as the conserved frequencies of a specific cell. We find that asymmetry does not impact the vibrational spectra, and the impact of sterols depends on the mobility of the components of the membrane. We demonstrate that vibrational spectra can be used to distinguish between membranes and, therefore, could be used in identification of different organisms. The method presented, here, can be immediately extended to other biological structures (e.g., amyloid fibers, polysaccharides, and protein-ligand structures) in order to fingerprint and understand vibrations of numerous biologically-relevant nanoscale structures.
-
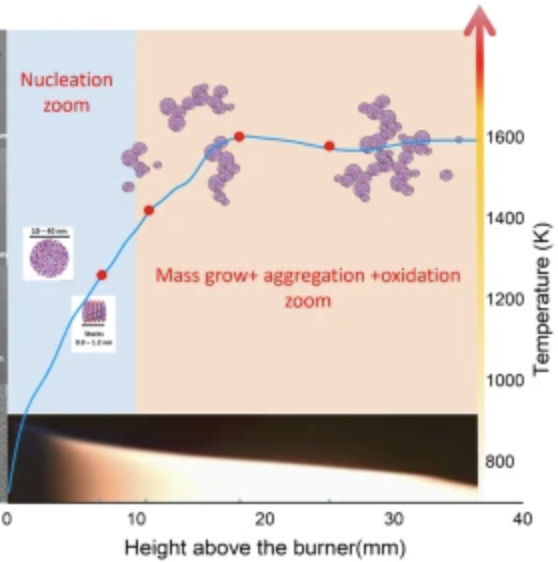 Elucidating the polycyclic aromatic hydrocarbons involved in soot inceptionCan Shao, Qi Wang, Wen Zhang, Anthony Bennett, Yang Li, and 5 more authorsCommun Chem, Oct 2023
Elucidating the polycyclic aromatic hydrocarbons involved in soot inceptionCan Shao, Qi Wang, Wen Zhang, Anthony Bennett, Yang Li, and 5 more authorsCommun Chem, Oct 2023Polycyclic aromatic hydrocarbons are the main precursors to soot particles in combustion systems. A lack of direct experimental evidence has led to controversial theoretical explanations for the transition from gas-phase species to organic soot clusters. This work focuses on sampling infant soot particles from well-defined flames followed by analysis using state-of-the-art mass spectrometry. We found that PAH molecules present in soot particles are all stabilomers. Kinetic Monte Carlo simulations and thermodynamic stability calculations further identify the detected PAHs as peri-condensed and without aliphatic chains. Van der Waals forces can easily link PAHs of such size and shape to form PAH dimers and larger clusters under the specified flame conditions. Our results provide direct experimental evidence that soot inception is initiated by a physical process under typical flame conditions. This work improves our understanding of aerosol particulates, which has implications for their environmental and climate change impacts.
- Exploring soot inception rate with stochastic modelling and machine learningLuke Di Liddo, Jacob C. Saldinger, Mehdi Jadidi, Paolo Elvati, Angela Violi, and 1 more authorCombustion and Flame, Oct 2023Special issue and Perspective on the Chemistry and Physics of Carbonaceious Particle Formation
A diverse range of polycyclic aromatic compounds (PACs) is thought to exist in flame environments before and during soot inception. This work seeks to develop a machine learning (ML)-based soot inception model that considers detailed and diverse PAC properties such as oxygenation, aliphatic content, radical character, size, and shape. To this end, temporal rates of change of PAC properties were computed by the stochastic modelling code SNapS2 and used as input to an ML model that predicts soot inception rate. The model is trained using experimentally-derived soot inception rates for three atmospheric pressure laminar premixed ethylene/air flames. An ML model (kernel ridge regression with a linear kernel) was developed to predict the soot inception rate in the three premixed flames. The soot inception rate predictions from this SNapS2-informed ML model outperformed the predictions from both the advanced soot modelling CFD code CoFlame and an ML model which used CFD-determined inputs (temperature and species concentrations). The final model had an R2 value of approximately 0.71 and a mean absolute error approximately 25% of the target values. The performance of the SNapS2-informed model suggests that detailed PAC properties are important to consider in inception modelling. While expanding this approach to other types of flames and fuels is crucial for future improvement to the model’s accuracy and generality, this methodology provides a successful framework for the current system. The success of this method demonstrates that ML can offer improvements in accuracy compared to current CFD inception models and the highlights the potential for ML in soot predictions.
-
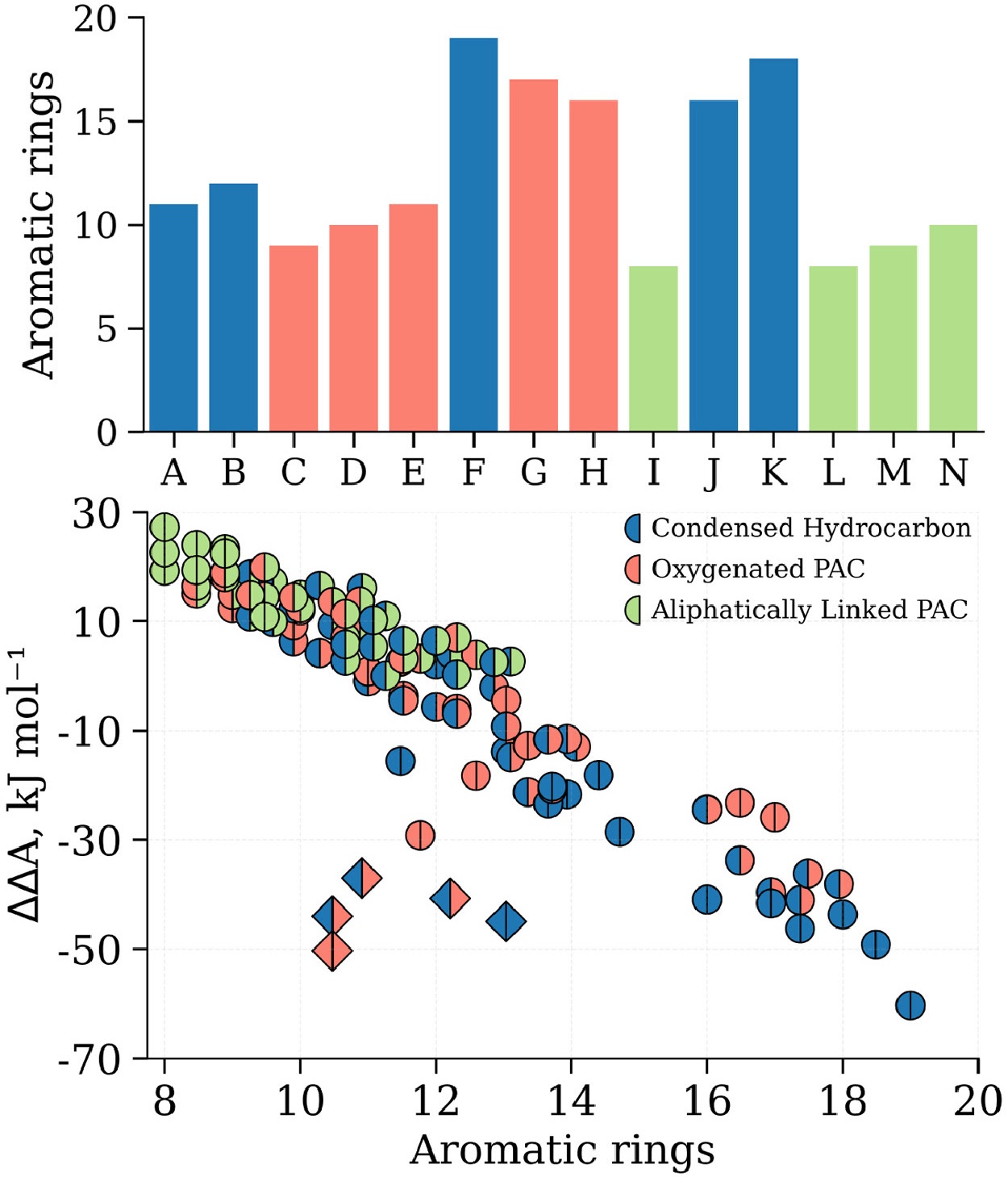 A machine learning framework to predict the aggregation of polycyclic aromatic compoundsJacob C. Saldinger, Paolo Elvati, and Angela VioliProceedings of the Combustion Institute, Oct 2023
A machine learning framework to predict the aggregation of polycyclic aromatic compoundsJacob C. Saldinger, Paolo Elvati, and Angela VioliProceedings of the Combustion Institute, Oct 2023The physical aggregation of polycyclic aromatic compounds (PACs) is a key step in soot inception. In this work, we set out to elucidate which molecular properties of PACs influence the physical growth process and develop a machine learning framework to quantitatively relate these features to the propensity of PACs to physically dimerize. To this end, we identify a pool of compounds with a diverse range of properties and create a dataset of PAC monomers along with their calculated free energies of dimerization, obtained via molecular dynamics simulations enhanced by well-tempered Metadynamics. We then demonstrate that a machine learning model based on the least absolute shrinkage and selection operator (Lasso) is able to quantitatively learn how molecular features contribute to physical aggregation and predict the free energy of dimerization for new pairs of molecules. Results show that our model is able to accurately determine the stability of dimers obtained from both homo- and hetero-molecular dimerization cases. Our approach provides also a data driven method to determine the molecular features most important to predicting the dimer stability. Indeed, we identified size, shape, oxygenation, and presence of rotatable bonds as the most influential characteristics of PACs that contribute to physical dimerization. This work highlights the molecular complexity of the PAC monomers that must be accounted for in order to accurately represent physical aggregation. We anticipate that our approach is key to modeling soot inception as it allows for the efficient prediction of dimerization propensity from easily calculable molecular features.
2022
-
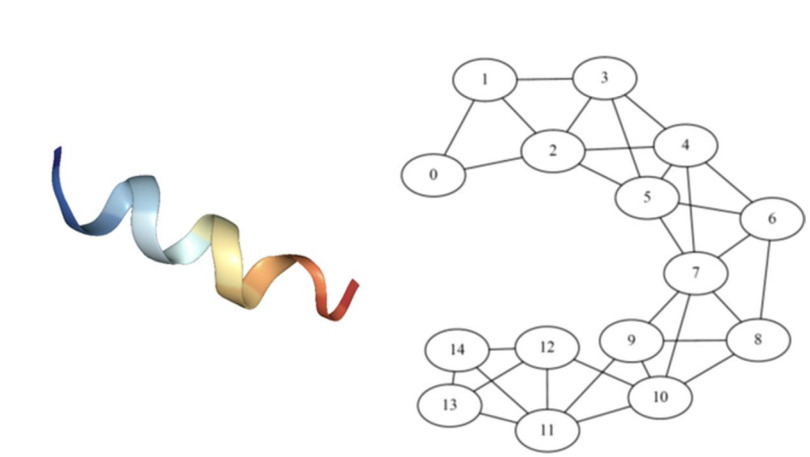 Struct2Graph: a graph attention network for structure based predictions of protein–protein interactionsAbram Baranwal, Jacob Saldinger, Emine S. Turali-emre, Paolo Elvati, Shivani Kozarekar, and 4 more authorsBMC Bioinformatics, Sep 2022
Struct2Graph: a graph attention network for structure based predictions of protein–protein interactionsAbram Baranwal, Jacob Saldinger, Emine S. Turali-emre, Paolo Elvati, Shivani Kozarekar, and 4 more authorsBMC Bioinformatics, Sep 2022Development of new methods for analysis of protein–protein interactions (PPIs) at molecular and nanometer scales gives insights into intracellular signaling pathways and will improve understanding of protein functions, as well as other nanoscale structures of biological and abiological origins. Recent advances in computational tools, particularly the ones involving modern deep learning algorithms, have been shown to complement experimental approaches for describing and rationalizing PPIs. However, most of the existing works on PPI predictions use protein-sequence information, and thus have difficulties in accounting for the three-dimensional organization of the protein chains.
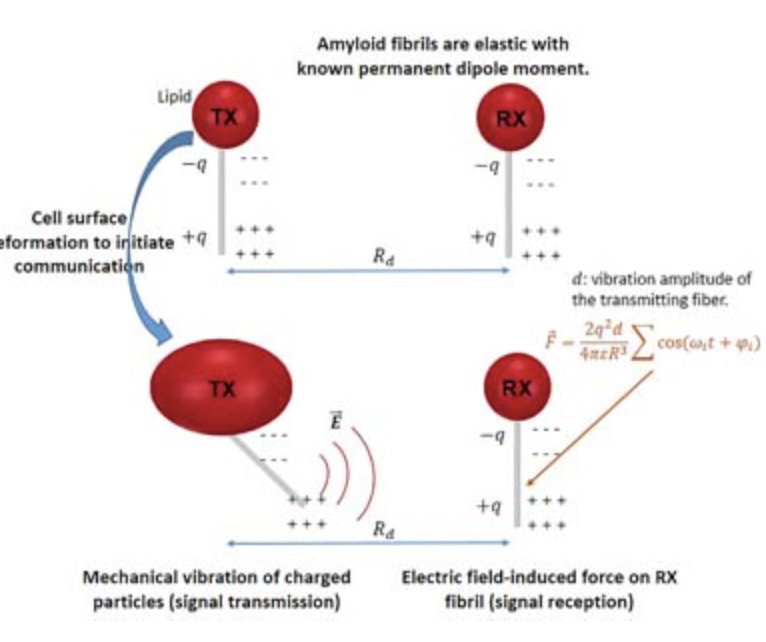
- A Multiphysics Modeling of Electromagnetic Signaling Phenomena at kHz-GHz Frequencies in Bacterial BiofilmsNavid Barani, Kamal Sarabandi, Nicholas A. Kotov, J. Scott Vanepps, Paolo Elvati, and 2 more authorsIEEE Access, Apr 2022
This paper presents a model that describes a possible mechanism for electromagnetic (EM) signal transmission and reception by bacterial cells within their biofilm communities. Bacterial cells in biofilms are embedded into a complex extracellular matrix containing, among other components, charged helical nanofibrils from amyloid-forming peptides. Based on the current knowledge about the nanoscale structure and dynamics of the amyloids, we explore a hypothetical model that the mechanical vibration of these nanofibrils allows the cells to transmit EM signals to their neighboring cells and the surrounding environment. For the reception, the induced electric field can either exert force on the charges of adjacent nanofibrils associated with the neighboring cells or affect the placement/conformation of a certain charged messenger protein within the cell. The proposed model is based on a coupled system of electrical and mechanical nanoscale structures, which predicts signal transmission and reception within kHz-GHz frequency ranges. Different mechanisms for generating EM signals at various frequency bands related to the structure of the cell and their biofilm constituents are discussed.
-
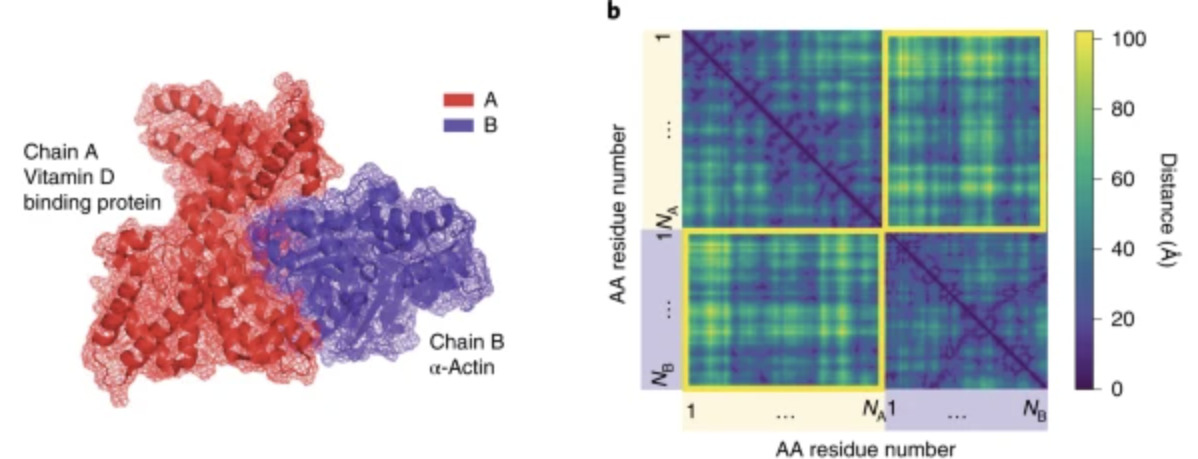 Unifying structural descriptors for biological and bioinspired nanoscale complexesMinjeong Cha, Emine Sumeyra Turali Emre, Xiongye Xiao, Ji-Young Kim, Paul Bogdan, and 3 more authorsNature Computational Science, Apr 2022
Unifying structural descriptors for biological and bioinspired nanoscale complexesMinjeong Cha, Emine Sumeyra Turali Emre, Xiongye Xiao, Ji-Young Kim, Paul Bogdan, and 3 more authorsNature Computational Science, Apr 2022Biomimetic nanoparticles are known to serve as nanoscale adjuvants, enzyme mimics and amyloid fibrillation inhibitors. Their further development requires better understanding of their interactions with proteins. The abundant knowledge about protein–protein interactions can serve as a guide for designing protein–nanoparticle assemblies, but the chemical and biological inputs used in computational packages for protein–protein interactions are not applicable to inorganic nanoparticles. Analysing chemical, geometrical and graph-theoretical descriptors for protein complexes, we found that geometrical and graph-theoretical descriptors are uniformly applicable to biological and inorganic nanostructures and can predict interaction sites in protein pairs with accuracy >80% and classification probability ~90%. We extended the machine-learning algorithms trained on protein–protein interactions to inorganic nanoparticles and found a nearly exact match between experimental and predicted interaction sites with proteins. These findings can be extended to other organic and inorganic nanoparticles to predict their assemblies with biomolecules and other chemical structures forming lock-and-key complexes.
-
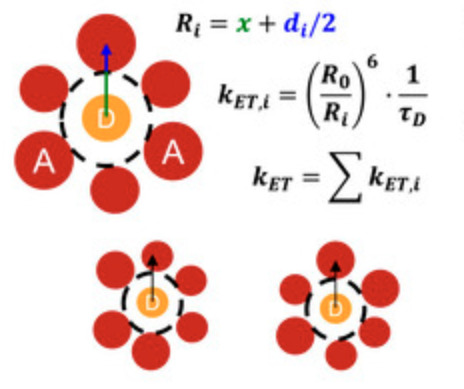 Distance-dependent resonance energy transfer in alkyl-terminated Si nanocrystal solidsZhaohan Li, Zachary L. Robinson, Paolo Elvati, Angela Violi, and Uwe R. KortshagenThe Journal of Chemical Physics, Mar 2022
Distance-dependent resonance energy transfer in alkyl-terminated Si nanocrystal solidsZhaohan Li, Zachary L. Robinson, Paolo Elvati, Angela Violi, and Uwe R. KortshagenThe Journal of Chemical Physics, Mar 2022Understanding and controlling the energy transfer between silicon nanocrystals is of significant importance for the design of efficient optoelectronic devices. However, previous studies on silicon nanocrystal energy transfer were limited because of the strict requirements to precisely control the inter-dot distance and to perform all measurements in air-free environments to preclude the effect of ambient oxygen. Here, we systematically investigate the distance-dependent resonance energy transfer in alkyl-terminated silicon nanocrystals for the first time. Silicon nanocrystal solids with inter-dot distances varying from 3 to 5 nm are fabricated by varying the length and surface coverage of alkyl ligands in solution-phase and gas-phase functionalized silicon nanocrystals. The inter-dot energy transfer rates are extracted from steady-state and time-resolved photoluminescence measurements, enabling a direct comparison to theoretical predictions. Our results reveal that the distance-dependent energy transfer rates in Si NCs decay faster than predicted by the Förster mechanism, suggesting higher-order multipole interactions.
- Structural characterization of PSMa1 functional amyloids in Staphylococcus aureus biofilmChloe Luyet, Paolo Elvati, Yichun Wang, Changjiang Liu, J. Scott VanEpps, and 2 more authorsBiophysical Journal, Feb 2022
-
 Comparison of the static structure factor at long wavelengths for a dusty plasma liquid and other liquidsVitaliy Zhuravlyov, J. Goree, Jack F. Douglas, Paolo Elvati, and Angela VioliPhys. Rev. E, Nov 2022
Comparison of the static structure factor at long wavelengths for a dusty plasma liquid and other liquidsVitaliy Zhuravlyov, J. Goree, Jack F. Douglas, Paolo Elvati, and Angela VioliPhys. Rev. E, Nov 2022Especially small values of the static structure factor \(S(k)\)at long wavelengths, i.e., small \(k\), were obtained in an analysis of experimental data for a two-dimensional dusty plasma in its liquid state. For comparison, an analysis of \(S(k)\)data was carried out for many previously published experiments with other liquids. This analysis indicates that the magnitude of \(S(k)\)at small \(k\)is typically in the range of 0.02–0.13. In contrast, the corresponding value for a dusty plasma liquid was found to be as small as 0.0139. Another basic finding for the dusty plasma liquid is that \(S(k)\)at small \(k\)generally increases with temperature, with its lowest value, noted above, occurring near the melting point. Simulations were carried out for the dusty plasma liquid, and their results are generally consistent with the experiment. Since a dusty plasma has a soft interparticle interaction, our findings support earlier theoretical suggestions that a useful design strategy for creating materials having exceptionally low values of \(S(0)\), so-called hyperuniform materials, is the use of a condensed material composed of particles that
-
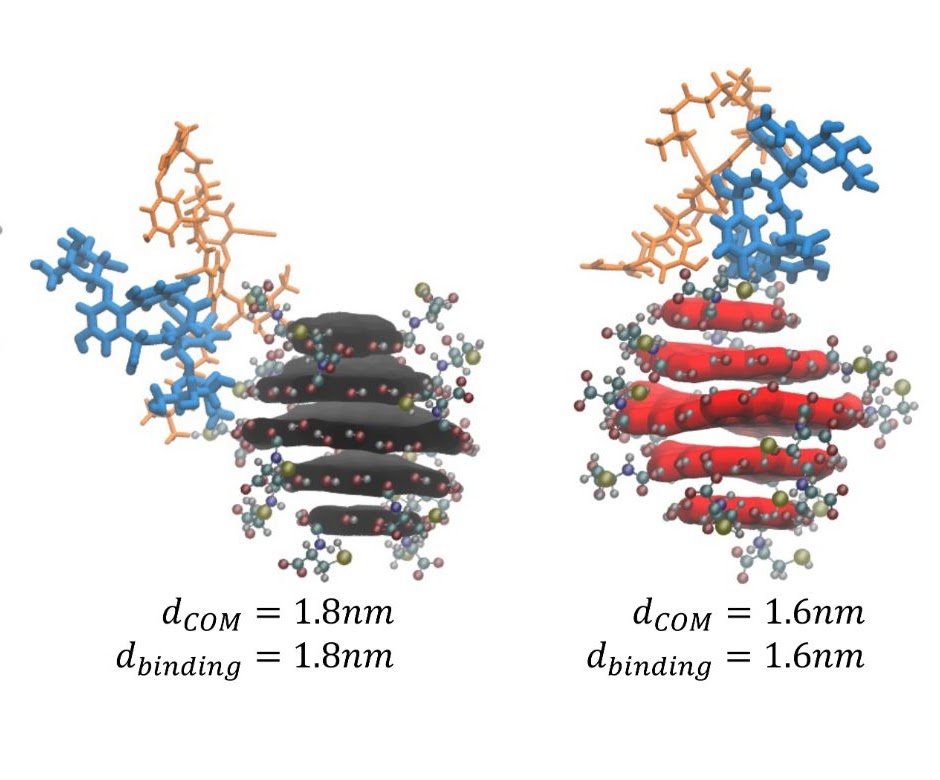 Chiral chromatography and surface chirality of carbon nanoparticlesMisché A. Hubbard, Chloe Luyet, Prashant Kumar, Paolo Elvati, J. Scott VanEpps, and 2 more authorsChirality, Nov 2022
Chiral chromatography and surface chirality of carbon nanoparticlesMisché A. Hubbard, Chloe Luyet, Prashant Kumar, Paolo Elvati, J. Scott VanEpps, and 2 more authorsChirality, Nov 2022Abstract Chiral carbon nanoparticles (CNPs) represent a rapidly evolving area of research for optical and biomedical technologies. Similar to small molecules, applications of CNPs as well as fundamental relationships between their optical activity and structural asymmetry would greatly benefit from their enantioselective separations by chromatography. However, this technique remains in its infancy for chiral carbon and other nanoparticles. The possibility of effective separations using high performance liquid chromatography (HPLC) with chiral stationary phases remains an open question whose answer can also shed light on the components of multiscale chirality of the nanoparticles. Herein, we report a detailed methodology of HPLC for successful separation of chiral CNPs and establish a path for its future optimization. A mobile phase of water/acetonitrile was able to achieve chiral separation of CNPs derived from L- and D-cysteine denoted as L-CNPs and D-CNPs. Molecular dynamics simulations show that the teicoplanin-based stationary phase has a higher affinity for L-CNPs than for D-CNPs, in agreement with experiments. The experimental and computational findings jointly indicate that chiral centers of chiral CNPs are present at their surface, which is essential for the multiple applications of these chiral nanostructures and equally essential for interactions with biomolecules and circularly polarized photons.
-
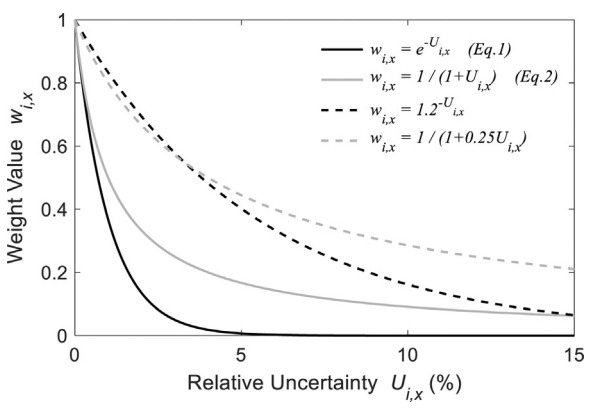 Uncertainty-based weight determination for surrogate optimizationDoohyun Kim and Angela VioliCombustion and Flame, Nov 2022
Uncertainty-based weight determination for surrogate optimizationDoohyun Kim and Angela VioliCombustion and Flame, Nov 2022Fuel surrogates are simplified models that mimic the combustion characteristics of very complex real transportation fuels and enable a detailed description of the computational system for the targeted real fuels. Current efforts in surrogate development focus on matching multiple target properties of the real fuel using numerical optimization of species compositions. A way to solve the multi-objective optimization problem is to employ the weighted-sum approach with arbitrarily assigned weights. In this paper, we propose a novel approach to reduce such arbitrariness by incorporating physical information into the surrogate optimization process, leveraging uncertainties from experimental measurements and mixture property predictions to determine the weight of each target property. The underlying principle of this approach is assigning low weight for properties with high uncertainty since a tight emulation of that property is not necessary. We propose formulations that convert relative uncertainties of each target property into weights for multi-objective surrogate optimization, which penalize target properties with high uncertainties. The method is flexible enough to accommodate not only multiple uncertainty sources, but also users’ preferences and the sensitivity of weights to uncertainty variations can be readily adjusted. The proposed method is applied to formulate surrogate mixtures for three reference target jet fuels (Jet-A POSF-10325, JP-8 POSF-10264, JP-5 POSF-10289), which have considerably different hydrocarbon compositions and properties. The results show that the surrogates developed by the uncertainty-based weight method emulate the target properties of fuels very closely, and they are able to capture the compositional characteristics of the target fuels as well.
2021
-
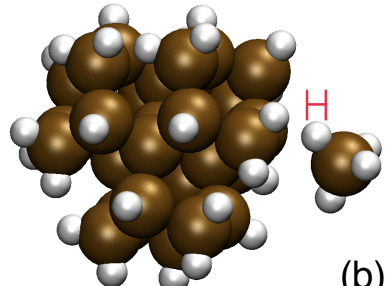 On the growth of Si nanoparticles in non-thermal plasma: physisorption to chemisorption conversionXuetao Shi, Paolo Elvati, and Angela VioliJournal of Physics D: Applied Physics, Jun 2021
On the growth of Si nanoparticles in non-thermal plasma: physisorption to chemisorption conversionXuetao Shi, Paolo Elvati, and Angela VioliJournal of Physics D: Applied Physics, Jun 2021Non-thermal plasma systems offer unique opportunities in the fields of bio-imaging, drug delivery, photovoltaics, microelectronics manufacturing. Such interests are largely inspired by the fact that hot plasma electrons coexist with neutral species and ions close to room-temperature under non-thermal plasma conditions. Modeling of these systems requires a deep understanding of the atomistic processes underlying the rich chemistry of the various radicals and ions with the nascent nanoparticle (NP) surface. A key parameter for determining the contribution of a certain radical/ion species to the NP surface growth, called sticking coefficient, is computed as a weighted sum from the simulated sticking outcomes with different collision velocities drawn from a Maxwell–Boltzmann distribution at certain temperatures. In this work, the collisions of SiH x (x\hspace0.167em=\hspace0.167em1–4) fragments and silicon cluster (Si4, Si2H6, and Si29H36) surfaces, responsible for the sticking coefficients, are simulated by molecular dynamics with a reactive force field. The dependence of sticking coefficients on temperature, H coverage of both silane fragments and cluster surfaces, and the size of the cluster, are systematically examined. And the mechanism underlying the sticking events, specifically the conversion of physical aggregation to chemisorption is investigated to better understand the complex interplay between factors influencing the surface growth. The detailed and multi-parameter model of sticking coefficients, accompanied by the mechanism study of physisorption to chemisorption conversion, provides a more accurate and robust approximation of surface growth rate using sticking coefficients, and a deeper understanding of surface growth processes, for the wider non-thermal plasma simulation community.
-
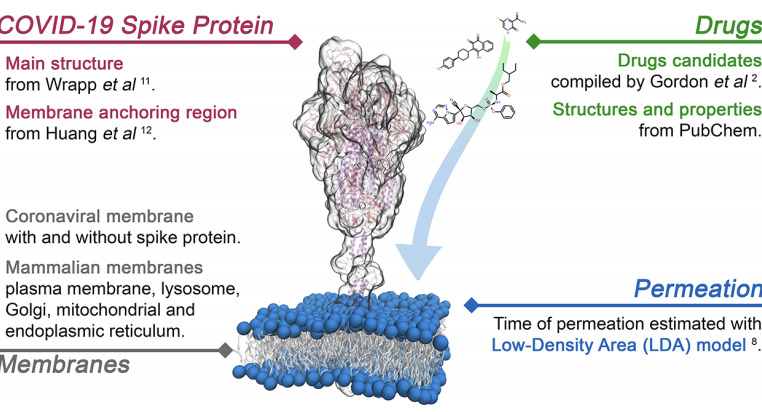 On Drug-Membrane Permeability of Antivirals for SARS-CoV-2Paolo Liu and Angela VioliThe Journal of Physical Chemistry Letters, Feb 2021
On Drug-Membrane Permeability of Antivirals for SARS-CoV-2Paolo Liu and Angela VioliThe Journal of Physical Chemistry Letters, Feb 2021One of the key parameters required to identify effective drugs is membrane permeability, as a compound intended for an intracellular target with poor permeability will have low efficacy. In this paper, we leverage a computational approach recently developed by our group to study the interactions between nanoparticles and mammalian membranes to study the time of entry of a variety of drugs into the viral envelope of coronavirus as well as cellular organelles. Using a combination of all-atoms molecular dynamics simulations and statistical analysis, we consider both drug characteristics and membrane properties to determine the behavior of 79 drugs and their interactions with the viral envelope, composed of the membrane and spike protein, as well as five other membranes that correspond to various mammalian compartments (lysosome, plasma, Golgi, mitochondrial, and endoplasmic reticulum membranes). The results highlight important trends that can be exploited for drug design, from the relatively high permeability of the viral envelope and the effect of transmembrane proteins, to the differences in permeability between organelles. When compared with bioavailability data present in the literature, the model results suggest a negative correlation between time of permeation and bioavailability of promising drugs. The method is general and flexible and can be employed for a variety of molecules, from small drugs to small nanoparticles, as well to a variety of biological membranes. Overall, the results indicate that this model can contribute to the identification of successful drugs as it predicts the ability of compounds to reach both intended and unintended intracellular targets.
-
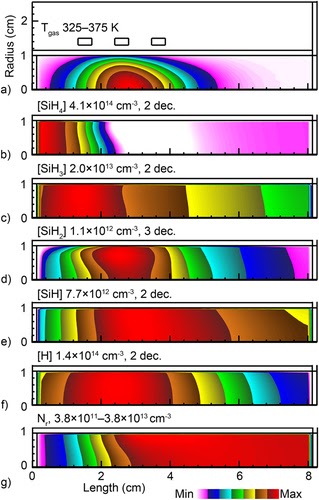 Scaling of silicon nanoparticle growth in low temperature flowing plasmasSteven J. Lanham, Jordyn Polito, Xuetao Shi, Paolo Elvati, Angela Violi, and 1 more authorJournal of Applied Physics, Oct 2021
Scaling of silicon nanoparticle growth in low temperature flowing plasmasSteven J. Lanham, Jordyn Polito, Xuetao Shi, Paolo Elvati, Angela Violi, and 1 more authorJournal of Applied Physics, Oct 2021Low temperature plasmas are an emerging method to synthesize high quality nanoparticles (NPs). An established and successful technique to produce NPs is using a capacitively coupled plasma (CCP) in cylindrical geometry. Although a robust synthesis technique, optimizing or specifying NP properties using CCPs, is challenging. In this paper, results from a computational investigation for the growth of silicon NPs in flowing inductively coupled plasmas (ICPs) using Ar/SiH4 gas mixtures of up to a few Torr are discussed. ICPs produce more locally constrained and quiescent plasma potentials. These positive plasma potentials produce an electrostatic trap for negatively charged NPs, which can significantly extend the residence time of NPs in the plasma, which in turn provides a controllable period for particle growth. The computational platforms used in this study consist of a two-dimensional plasma hydrodynamics model, a three-dimensional nanoparticle growth and trajectory tracking model, and a molecular dynamics simulation for deriving reactive sticking coefficients of silane radicals on Si NPs. Trends for the nanoparticle growth as a function of SiH4 inlet fraction, gas residence time, energy deposition per particle, pressure, and reactor diameter are discussed. The general path for particle synthesis is the trapping of small NPs in the positive electrostatic potential, followed by entrainment in the gas flow upon reaching a critical particle size. Optimizing or controlling NP synthesis then depends on the spatial distribution of plasma potential, the density of growth species, and the relative time that particles spend in the electrostatic trap and flowing through higher densities of growth species upon leaving the trap.
-
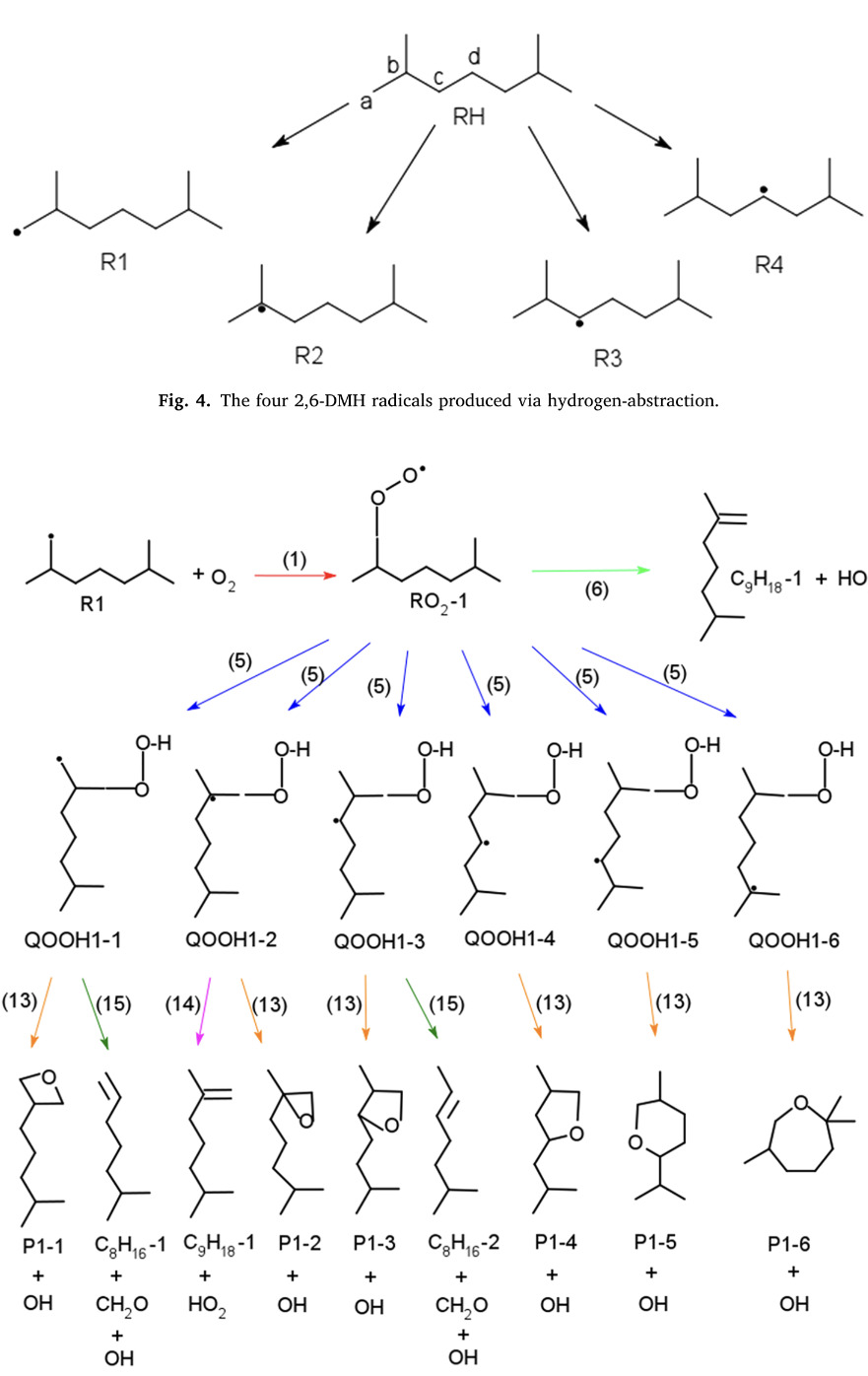 Oxidation of 2,6-dimethylheptane at low temperature: Kinetic modeling and experimental studyTanjin He, Doohyun Kim, Tyler Dillstrom, Kaiyuan Cai, Peng Zhang, and 4 more authorsFuel, Oct 2021
Oxidation of 2,6-dimethylheptane at low temperature: Kinetic modeling and experimental studyTanjin He, Doohyun Kim, Tyler Dillstrom, Kaiyuan Cai, Peng Zhang, and 4 more authorsFuel, Oct 2021Branched alkanes represent an important class of compounds in conventional fuels and some bio-derived fuels. This study is dedicated to the investigation of the low-temperature oxidation chemistry of 2,6-dimethylheptane using a combination of experimental and computational methods. All the reactants, transition states, and products in the first oxidation stage, which are crucial to the initiation reactions in the low-temperature reaction chain, were optimized through the B3LYP/CBSB7 level of theory and a kinetic mechanism that included the new reaction pathways was assembled. Ignition delay time measurements were carried out in a rapid compression machine and the results were compared with modeling predictions. The kinetic mechanism is able to capture both the first and total ignition delay times with a root-mean-square deviation of 39.6%. In addition, sensitivity analysis is performed to quantify the impact of newly developed chemistry of 2,6-dimethylheptane on ignition delay time. Rate parameters found in this study may be applicable to other branched alkanes with similar molecular structure.
-
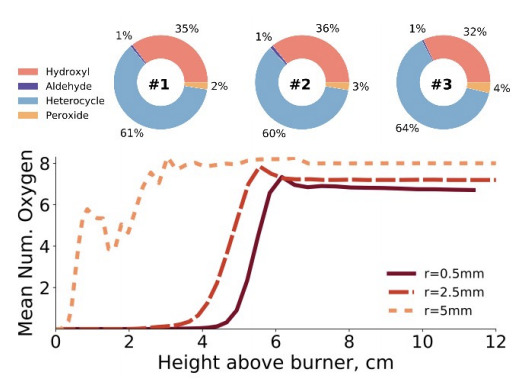 Stochastic and network analysis of polycyclic aromatic growth in a coflow diffusion flameJacob C. Saldinger, Paolo Elvati, and Angela VioliPhys. Chem. Chem. Phys., Oct 2021
Stochastic and network analysis of polycyclic aromatic growth in a coflow diffusion flameJacob C. Saldinger, Paolo Elvati, and Angela VioliPhys. Chem. Chem. Phys., Oct 2021An important step in predicting the growth of soot nanoparticles is understanding how gas phase variations affect the formation of their aromatic precursors. Once formed, these aromatic structures begin to assemble into nanoparticles and, regardless of the clustering process, the molecular properties of the aromatic precursors play an important role. Leveraging existing experimental data collected from a coflow Jet A-1 surrogate diffusion flame, in this paper we report on a detailed study of the spatial evolution of molecular structures of polycyclic aromatic compounds (PACs) and their corresponding formation pathways. To this end, we employed the SNapS2 kinetic Monte Carlo software to simulate the chemical evolution of PACs along multiple streamlines. The results show that growth only occurs along streamlines that traverse regions of high acetylene concentrations in the center of the flame. The PACs predicted in various conditions show diverse chemical properties, including aliphatic chains, five-membered, and heteroaromatic rings. PACs in streamlines close to the flame wings begin growing immediately due to the high temperature and large amounts of radical species, while PACs originating along inner streamlines do not appreciably grow until they pass through an area characterized by high radical concentrations. Using graph theory and network analysis, we investigated the complex reaction network generated by SNapS2 and determined that the growth pathways of many PACs center around a few stable structures that also promote oxygen addition reactions due to their morphology and long lifetimes. These pathways play a more significant role along streamlines near the centerline, compared to the flame wings, which show more variety due to the highly reactive environment encountered during early growth. The results of this study provide insights on the reaction pathways that determine the properties of PACs at different flame locations as well as information on the chemical characteristics of the formed PACs, with emphasis on oxygenated structures.
-
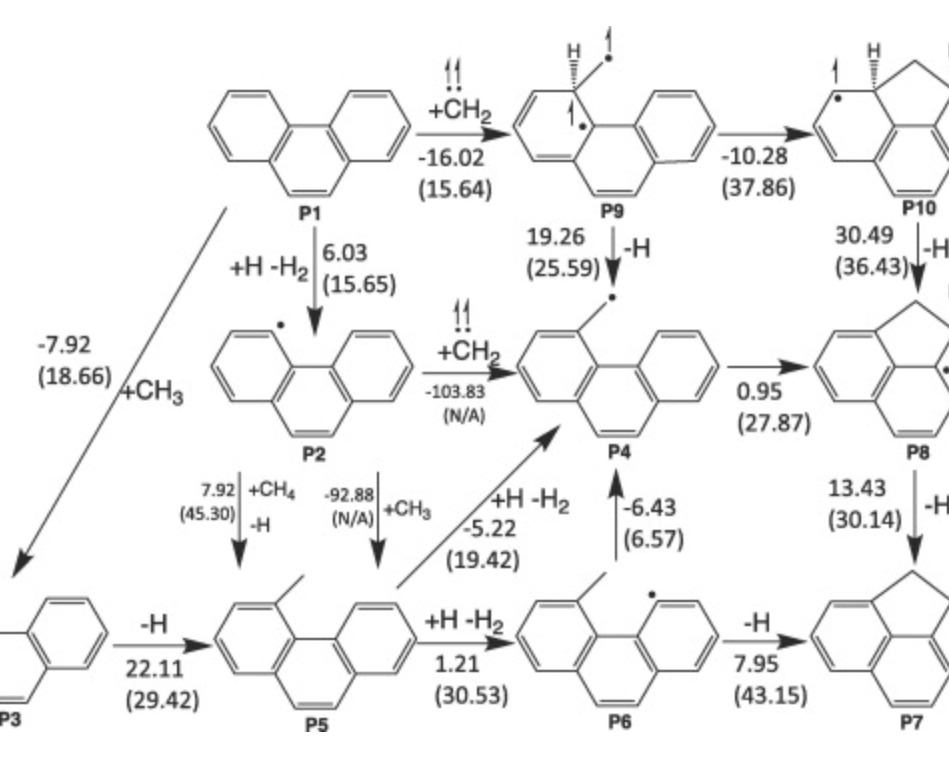 Reaction pathways for the formation of five-membered rings onto polyaromatic hydrocarbon frameworkXuetao Shi, Qi Wang, and Angela VioliFuel, Oct 2021
Reaction pathways for the formation of five-membered rings onto polyaromatic hydrocarbon frameworkXuetao Shi, Qi Wang, and Angela VioliFuel, Oct 2021Experimental and theoretical evidences accumulated over the years have highlighted the role of polycyclic aromatic hydrocarbons as molecular precursors to soot particles. However, many of their physical and chemical characteristics are still under debate, as well as the mechanisms that drive their transition from gaseous species to solid carbonaceous particles. The formation of five-membered rings can be described with three types of reactive sites present on hydrocarbons: (1) a “free-edge” reactive site and a C3 gas phase species (C2 + C3), (2) a “zig-zag” site and a C2 gas phase species (C3 + C2), and (3) an “armchair” site and a C1 gas phase species (C4 + C1). In this work, we focus our attention on the last two categories and use ab initio G3-type electronic structure calculations to explore systematically possible reaction pathways leading to the formation of five-membered rings. Specifically, our study reports on reaction pathways leading to the formation of acenaphthene and acenaphthylene starting from the zig-zag type site on naphthalene and to the formation of five-membered ring in the form of fluorene and 4H-cyclopenta[def]phenanthrene starting from the “armchair” site on biphenyl or phenanthrene. The relative importance of the new reaction pathways has been investigated in a 0-D Closed Homogeneous Batch Reactor, where the conditions are taken from experimental data. Furthermore, the new reaction pathways, together with temperature dependent rate constants, provide a more complete description of the formation of embedded five-membered rings, such as methylene-bridged PAHs identified in catechol pyrolysis, that can play an important role not only in the gas-phase chemistry of aromatics but also as pathways to five-membered rings on armchair sites and cross-linking for soot models.
-
 Combustion by-products and their health effects: Summary of the 16th international congressAngela Violi, Stephania Cormier, Brian Gullett, Stina Jansson, Slawo Lomnicki, and 3 more authorsFuel, Oct 2021
Combustion by-products and their health effects: Summary of the 16th international congressAngela Violi, Stephania Cormier, Brian Gullett, Stina Jansson, Slawo Lomnicki, and 3 more authorsFuel, Oct 2021The 16th International Congress on Combustion By-Products and their Health Effects (PIC2019) was held in Ann Arbor, Michigan, from July 10 to 12, 2019. For the last 28 years, this conference has served as an interdisciplinary platform for the discussion of the formation, environmental fate, health effects, policy, and remediation of combustion by-products. The technical areas for PIC2019 included mobile and stationary sources in urban environments, open fires, in- door air pollution, and halogenated pollutants. The congress was sponsored by the National Institute of Environmental Health Sciences (NIEHS), the U.S. EPA, the School of Public Health at the University of Michigan, the Civil and Environmental Engineering Department at the University of Michigan, the Mechanical Engineering Department at the University of Michigan, the Aerospace Engineering Department at the University of Michigan, and the Climate and Space Sciences and Engineering Department at the University of Michigan. Special features of the conference included a career path and round table discussion on translating research and engaging communities.
-
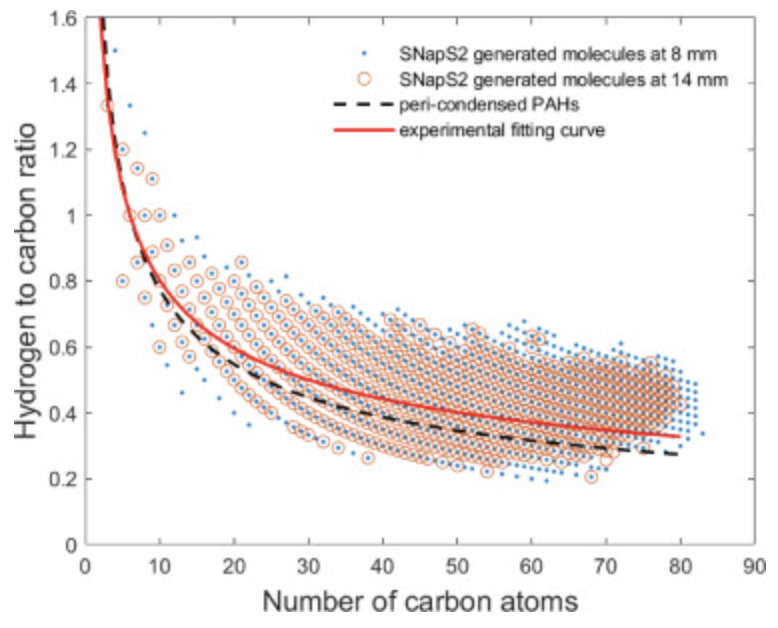 Molecular structures in flames: A comparison between SNapS2 and recent AFM resultsQi Wang, Jacob C. Saldinger, Paolo Elvati, and Angela VioliProceedings of the Combustion Institute, Oct 2021
Molecular structures in flames: A comparison between SNapS2 and recent AFM resultsQi Wang, Jacob C. Saldinger, Paolo Elvati, and Angela VioliProceedings of the Combustion Institute, Oct 2021Models capturing the growth of polycyclic aromatic compounds (PACs) and nanoparticles in combustion have always faced a large degree of uncertainty due to the paucity of detailed direct experimental validation. In particular, data on molecular structures, chemical composition, size, cross-linking, and aliphatic chains is still very limited. In the past few years, we have developed an atomistic code, SNapS2, that models the formation of PACs in combustion conditions, providing information on the chemical and structural evolution of these compounds. In this paper, we present a detailed analysis of the compounds formed in a premixed ethylene-air flame that was previously characterized experimentally by AFM (Commodo et al. 2019). The comparison is based on molecular structures and extends to other properties of PACs. The results demonstrate that SNapS2 is able to capture the vast array of PACs in terms of a large variety of functional groups. The simulations, confirmed to a large extent by the experimental data, show the presence of different types of oxygenated (e.g., phenol, ketone) and cyclic (acenaphthylene-type, acenaphthene-type, and fluorene-type five-membered rings) functional groups. Moreover, the structures that were not observed in the simulations (indane-type five-membered rings or six-membered rings containing oxygen) are missing due to the lack of formation pathways, highlighting the fact that important kinetic mechanisms are still missing in the literature. Finally, we observed that a preponderant number of high molecular mass PACs are curved, which may have an impact on their aggregation and sampling. Overall, while some discrepancies remain due to the inherent limitation of the model and the AFM techniques, this work demonstrates the unique capabilities of the SNapS2 code to provide insights on the structures and chemical pathways of PACs.
2020
- On sparse identification of complex dynamical systems: A study on discovering influential reactions in chemical reaction networksFarshad Harirchi, Doohyun Kim, Omar Khalil, Sijia Liu, Paolo Elvati, and 3 more authorsFuel, Nov 2020
-
 Molecular Dynamics Study of Dehydrated Lipopolysaccharide MembraneChangjiang Liu, Paolo Elvati, and Angela ViolibioRxiv, Nov 2020
Molecular Dynamics Study of Dehydrated Lipopolysaccharide MembraneChangjiang Liu, Paolo Elvati, and Angela ViolibioRxiv, Nov 2020The outer membrane of bacteria is known to play an important role in the rapid response to desiccation, although the causes and the extent of these effects are still mostly unclear. For this reason, in this work we study the desiccation response of the Gram-negative lipopolysaccha-ride (LPS) bacterial outer membranes. By analyzing molecular dynamics simulations of LPS membranes of different composition during desiccation, we identified the formation of a rigid protective layer of polysaccharides that not only reduces the water evaporation but is also able to indirectly preserve several structural features and as such membrane functionality. Overall, we found that the presence of polysaccharide layer is critical in the conservation of a layer of water in proximity of the hydrophobic region as well as the structure of the lipids acyl chain structure.Competing Interest StatementThe authors have declared no competing interest.
-
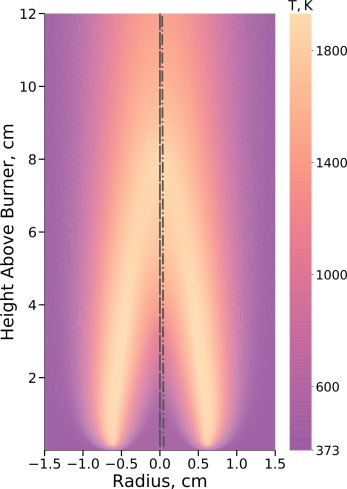 Characterizing the diversity of aromatics in a coflow diffusion Jet A-1 surrogate flameJacob C. Saldinger, Qi Wang, Paolo Elvati, and Angela VioliFuel, Nov 2020
Characterizing the diversity of aromatics in a coflow diffusion Jet A-1 surrogate flameJacob C. Saldinger, Qi Wang, Paolo Elvati, and Angela VioliFuel, Nov 2020Understanding the formation of soot precursors requires properly taking into account their diverse structure and chemistry. To study the extent of this variety in a realistic combustion system, we simulated the growth of polycyclic aromatic compounds (PACs) in a diffusion coflow flame using a three-component Jet A-1 surrogate (n-decane/ propylbenzene/ propylcyclohexane) as fuel. A kinetic Monte Carlo software, which generates samples of the atomistic evolution of gas-phase molecules’ growth, was used to characterize the PACs’ growth in this flame. We leveraged our simulations to identify a number of aromatic structures. In addition to aromatic hydrocarbons, we were able to reproduce the diverse array of oxygenated PACs observed experimentally between 150u and 450u, specifically a number of PACs with five-membered carbon rings and furan, hydroxl, and ketone groups. The amount of PACs with masses between 450u and 600u was slightly over-predicted, indicating that in this mass range there are possibly other mechanisms (e.g., radical-radical interactions) that reduce the concentration of these PACs. As our growth mechanism is focused mostly on aromatic growth, we did not observe the more aliphatic molecules seen in experiment. Overall, our results suggest that there is a wide range of structures with different degrees of oxygenation and aromaticity that must be accounted for when modeling PACs’ growth in combustion environments.
-
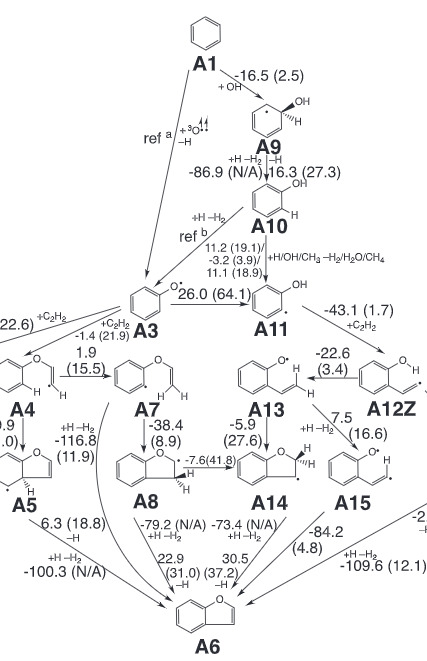 Chemical pathways for the formation of benzofuran and dibenzofuran in combustionXuetao Shi, Qi Wang, and Angela VioliCombustion and Flame, Nov 2020
Chemical pathways for the formation of benzofuran and dibenzofuran in combustionXuetao Shi, Qi Wang, and Angela VioliCombustion and Flame, Nov 2020Understanding and predicting the formation of polycyclic aromatic compounds (PACs) and their role in the formation of high molecular mass compounds is still an unresolved topic in combustion. PACs characteristics, such as chemical composition, size, and presence of side chains, play an important role not only in terms of environmental and health impact, but also when developing models that describe the formation of nanoparticles and soot. In this paper, we report on a detailed analysis of the reaction pathways describing the chemistry of furan-embedded PACs using ab initio G3-type electronic structure calculations leading to the formation of benzofuran and dibenzofuran from benzene and biphenyl. The 82 elementary reactions, identified in this work, contain unexplored pathways involving triplet oxygen atom and hydroxyl radical addition reactions. A protocol for improving the calculations of reaction energetics from ab initio compound methods is proposed, which consists of the thorough usage of IRCmax scheme to identify the transition state structure and an energy correction ( 0.2 kcal/mol) to the empirical term in G3 formula for systems with open-shell singlet type of electronic configurations. Based on these ab initio calculations, temperature dependent reaction rate constants are calculated according to statistical theories, together with thermodynamics data. Branching ratio analysis based on steady-state approximation is carried out to illustrate the relative importance of the new pathways in an ethylene premixed flame. Results show that the newly discovered benzofuran formation pathways can play a relative important role when in presence of phenol or phenoxyl radicals at various locations in the flame.
-
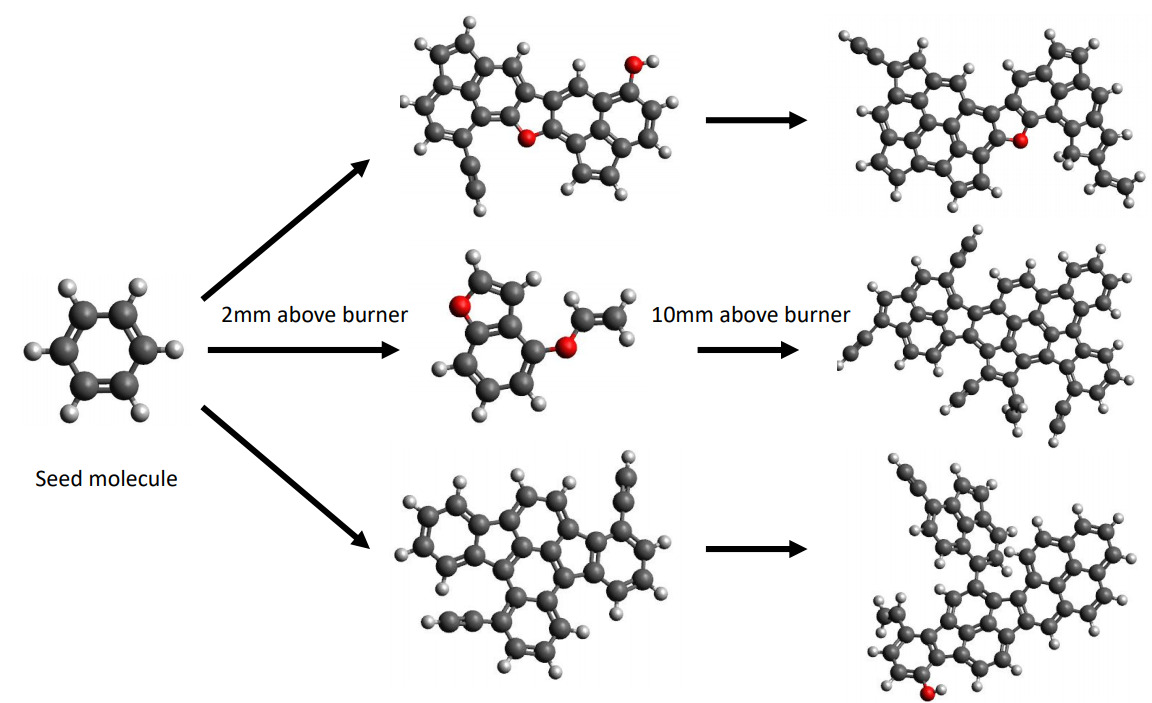 Insights on the effect of ethanol on the formation of aromaticsQi Wang, Jacob C. Saldinger, Paolo Elvati, and Angela VioliFuel, Nov 2020
Insights on the effect of ethanol on the formation of aromaticsQi Wang, Jacob C. Saldinger, Paolo Elvati, and Angela VioliFuel, Nov 2020For decades, ethanol has played an important role as a biofuel, as its addition to hydrocarbon fuels has been associated with a reduction in soot formation. The chemical mechanisms behind this phenomenon, however, are still not clear. In this paper, we contribute to this research area by elucidating the mechanisms that participate in the formation of polycyclic aromatic compounds (PACs) in an ethanol-doped ethylene flame, using a combination of deterministic and stochastic computational techniques. We specifically focus on the formation of oxygenated PACs and the chemical interactions of pure hydrocarbons and oxygen in six ethylene/air premixed flames with similar temperature profiles but different equivalence ratios and ethanol doping percentages. Our simulations confirm that an increase in the ethanol content results in a reduction of the formation of acetylene, small aromatics, and large PAHs. At the same time, the number of oxygenated PACs reach a maximum at a height above burner around 2–3 mm where they constitute 45% of all PACs: most of the oxygenated structures are phenols, mixed with approximately 15% of furans, and a small amount of ethers. Overall, the results indicate that the formation pathways of oxygenated PACs can compete with pure hydrocarbon growth mechanisms in the rapid growth region up to a height above burner of 2–3 mm. The rate of PACs’ growth then gradually slows down, with pure hydrocarbon growth mechanisms being the main contributors in this region of the flame.
- Chemical kinetic modeling study of methyl esters oxidation: Improvement on the prediction of early CO2 formationYi Zhou, Yunhua Gan, and Xiaolong GouFuel, Nov 2020
The early CO2 formation is a characteristic for the methyl esters group of biodiesel. A new reaction pathway was added to skeletal methyl esters mechanism for improving the prediction of early CO2 formation. The methyl decanoate, methyl 9-decenoate, methyl 5-decenoate and methyl stearate sub-mechanisms with added reaction pathway were optimized by adjusting reaction rate constants for more accurate prediction. Based on decoupling methodology, a new skeletal mechanism for methyl butanoate was constructed by integrating detailed H2/CO/C1 sub-mechanism, reduced C2-C3 sub-mechanism and methyl butanoate sub-mechanism. These improved mechanisms were validated well in a shock tube for ignition delay times and in a jet-stirred reactor for major species concentrations over wide operating conditions, respectively. When compared to available mechanism in the literature, the present mechanism has good improvement for the prediction of early CO2 formation. Moreover, the effect of newly added reactions on ignition delay times was analyzed by sensitivity analysis method. Added reaction of Fuel Radical = ME2J + Short Chain Hydrocarbon mainly causes the influence on ignition delay time at high temperature, and decrease the reactivity of oxidation of fuel radical. The reaction of OCHO + M<=>H + CO2 + M dominates the early CO2 formation, and makes less contribution to production of CO2 with higher temperature. The improved mechanisms, which consist of methyl esters from a relatively short to long carbon chain, have a good performance for the prediction of early CO2 formation.
2019
-
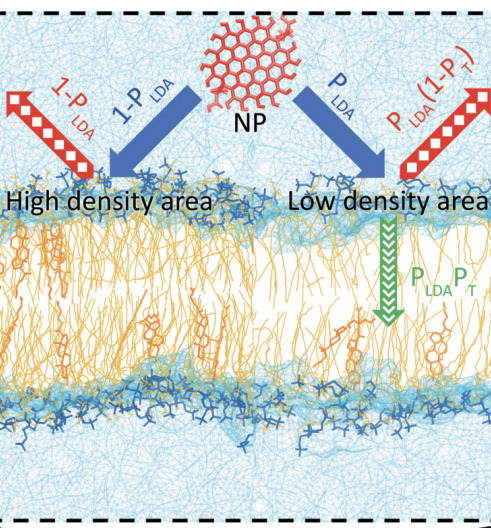 Predicting the Time of Entry of Nanoparticles in Lipid MembranesChangjiang Liu, Paolo Elvati, Sagardip Majumder, Yichun Wang, Allen P. Liu, and 1 more authorACS Nano, Sep 2019
Predicting the Time of Entry of Nanoparticles in Lipid MembranesChangjiang Liu, Paolo Elvati, Sagardip Majumder, Yichun Wang, Allen P. Liu, and 1 more authorACS Nano, Sep 2019The number of engineered nanoparticles for applications in the biomedical arena has grown tremendously over the last years due to advances in the science of synthesis and characterization. For most applications, the crucial step is the transport through a physiological cellular membrane. However, the behavior of nanoparticles in a biological matrix is a very complex problem that depends not only on the type of nanoparticle but also on its size, shape, phase, surface charge, chemical composition, and agglomeration state. In this paper, we introduce a streamlined theoretical model that predicts the average time of entry of nanoparticles in lipid membranes, using a combination of molecular dynamics simulations and statistical approaches. The model identifies four parameters that separate the contributions of nanoparticle characteristics (i.e., size, shape, solubility) from the membrane properties (density distribution). This factorization allows the inclusion of data obtained from both experimental and computational sources, as well as a rapid estimation of large sets of permutations in membranes. The robustness of the model is supported by experimental data carried out in lipid vesicles encapsulating graphene quantum dots as nanoparticles. Given the high level of interest across multiple areas of study in modulating intracellular targets, and the need to understand and improve the applications of nanoparticles and to assess their effect on human health (i.e., cytotoxicity, bioavailability), this work contributes to the understanding and prediction of interactions between nanoparticles and lipid membranes.
-
 Anti-Biofilm Activity of Graphene Quantum Dots via Self-Assembly with Bacterial Amyloid ProteinsYichun Wang, Usha Kadiyala, Zhibei Qu, Paolo Elvati, Christopher Altheim, and 3 more authorsACS Nano, Apr 2019
Anti-Biofilm Activity of Graphene Quantum Dots via Self-Assembly with Bacterial Amyloid ProteinsYichun Wang, Usha Kadiyala, Zhibei Qu, Paolo Elvati, Christopher Altheim, and 3 more authorsACS Nano, Apr 2019Bacterial biofilms represent an essential part of Earth’s ecosystem that can cause multiple ecological, technological, and health problems. The environmental resilience and sophisticated organization of biofilms are enabled by the extracellular matrix that creates a protective network of biomolecules around the bacterial community. Current anti-biofilm agents can interfere with extracellular matrix production but, being based on small molecules, are degraded by bacteria and rapidly diffuse away from biofilms. Both factors severely reduce their efficacy, while their toxicity to higher organisms creates additional barriers to their practicality. In this paper, we report on the ability of graphene quantum dots to effectively disperse mature amyloid-rich Staphylococcus aureus biofilms, interfering with the self-assembly of amyloid fibers, a key structural component of the extracellular matrix. Mimicking peptide-binding biomolecules, graphene quantum dots form supramolecular complexes with phenol-soluble modulins, the peptide monomers of amyloid fibers. Experimental and computational results show that graphene quantum dots efficiently dock near the N-terminus of the peptide and change the secondary structure of phenol-soluble modulins, which disrupts their fibrillation and represents a strategy for mitigation of bacterial communities.
-
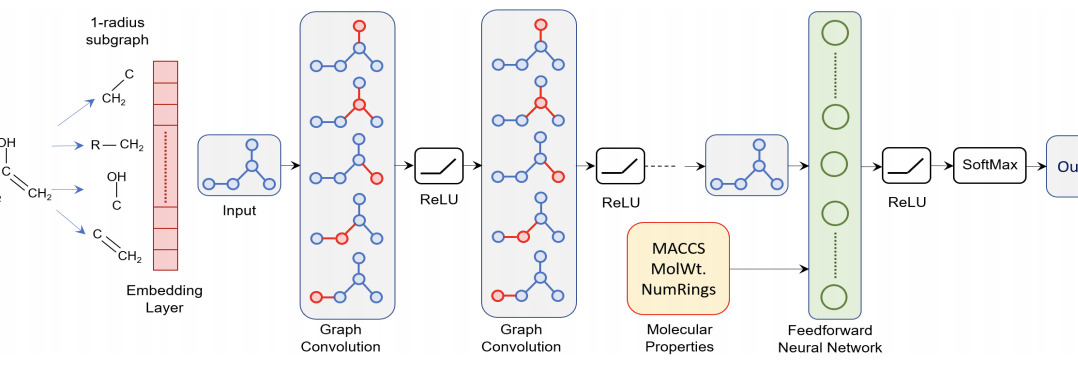 A deep learning architecture for metabolic pathway predictionMayank Baranwal, Abram Magner, Paolo Elvati, Jacob Saldinger, Angela Violi, and 1 more authorBioinformatics, Dec 2019
A deep learning architecture for metabolic pathway predictionMayank Baranwal, Abram Magner, Paolo Elvati, Jacob Saldinger, Angela Violi, and 1 more authorBioinformatics, Dec 2019Understanding the mechanisms and structural mappings between molecules and pathway classes are critical for design of reaction predictors for synthesizing new molecules. This article studies the problem of prediction of classes of metabolic pathways (series of chemical reactions occurring within a cell) in which a given biochemical compound participates. We apply a hybrid machine learning approach consisting of graph convolutional networks used to extract molecular shape features as input to a random forest classifier. In contrast to previously applied machine learning methods for this problem, our framework automatically extracts relevant shape features directly from input SMILES representations, which are atom-bond specifications of chemical structures composing the molecules.Our method is capable of correctly predicting the respective metabolic pathway class of 95.16\\% of tested compounds, whereas competing methods only achieve an accuracy of 84.92\\% or less. Furthermore, our framework extends to the task of classification of compounds having mixed membership in multiple pathway classes. Our prediction accuracy for this multi-label task is 97.61\\%. We analyze the relative importance of various global physicochemical features to the pathway class prediction problem and show that simple linear/logistic regression models can predict the values of these global features from the shape features extracted using our framework.https://github.com/baranwa2/MetabolicPathwayPrediction.Supplementary data are available at Bioinformatics online.
-
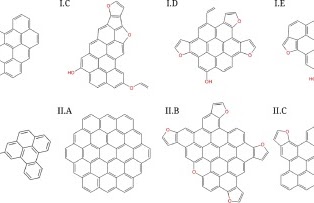 The role of molecular properties on the dimerization of aromatic compoundsPaolo Elvati, Kirk Turrentine, and Angela VioliProceedings of the Combustion Institute, Dec 2019
The role of molecular properties on the dimerization of aromatic compoundsPaolo Elvati, Kirk Turrentine, and Angela VioliProceedings of the Combustion Institute, Dec 2019Recent results have shown the presence and importance of oxygen chemistry during the growth of aromatic compounds, leading to the formation of oxygenated structures that have been identified in various environments. Since the formation of polycyclic aromatic compounds (PAC) bridge the formation of gas-phase species with particle inception, in this work we report a detailed analysis of the effects of molecular characteristics on physical growth of PAC via dimerization. We have included oxygen content, mass, type of bonds (rigid versus rotatable), and shape as main properties of the molecules and studied their effect on the propensity of these structures to form homo-molecular and hetero-molecular dimers. Using enhanced sampling molecular dynamics techniques, we have computed the free energy of dimerization in the temperature range 500–1680 K. Initial structures used in this study were obtained from experimental data. The results show that the effects of shape, presence of oxygen, mass, and internal bonds are tightly intertwined, and that their relative importance changes with temperature. In general, mass and the presence of rotatable bonds are the most influential factor to predict dimerization propensity among the one considered. The results provide knowledge on the inception step and the role that particle characteristics play during inception. In addition, our study highlights the fact that current models that use stabilomers as monomers for physical aggregation are overestimating the importance of their dimerization during particle nucleation.
- A New Data-Driven Sparse-Learning Approach to Study Chemical Reaction NetworksFarshad Harirchi, Doohyun Kim, Omar A. Khalil, Sijia Liu, Paolo Elvati, and 2 more authorsDec 2019
Chemical kinetic mechanisms can be represented by sets of elementary reactions that are easily translated into mathematical terms using physicochemical relationships. The schematic representation of reactions captures the interactions between reacting species and products. Determining the minimal chemical interactions underlying the dynamic behavior of systems is a major task. In this paper, we introduce a novel approach for the identification of the influential reactions in chemical reaction networks for combustion applications, using a data-driven sparse-learning technique. The proposed approach identifies a set of influential reactions using species concentrations and reaction rates, with minimal computational cost without requiring additional data or simulations. The new approach is applied to analyze the combustion chemistry of H2 and C3H8 in a constant-volume homogeneous reactor. The influential reactions identified by the sparse-learning method are consistent with the current kinetics knowledge of chemical mechanisms. Additionally, we show that a reduced version of the parent mechanism can be generated as a combination of the influential reactions identified at different times and conditions and that for both H2 and C3H8 this reduced mechanism performs closely to the parent mechanism as a function of ignition delay over a wide range of conditions. Our results demonstrate the potential of the sparse-learning approach as an effective and efficient tool for mechanism analysis and mechanism reduction.
-
 The effect of molecular structures of alkylbenzenes on ignition characteristics of binary n-heptane blendsDongil Kang, Doohyun Kim, Kwang Hee Yoo, Angela Violi, and André BoehmanProceedings of the Combustion Institute, Dec 2019
The effect of molecular structures of alkylbenzenes on ignition characteristics of binary n-heptane blendsDongil Kang, Doohyun Kim, Kwang Hee Yoo, Angela Violi, and André BoehmanProceedings of the Combustion Institute, Dec 2019Alkylbenzenes are major aromatic constituents of real transportation fuels and important surrogate components. In this study, the structural impact of nine alkylbenzenes on their ignition characteristics is experimentally and computationally investigated with particular emphasis on the blending effect with significantly more reactive normal alkanes. Experimental comparisons of mono-alkylbenzenes (toluene, ethylbenzene, n-propylbenzene, iso-propylbenzene) from a modified CFR engine showed that the difference in pure alkylbenzene reactivity significantly diminished when blended with n-heptane, as the strength of the radical scavenging effect of all three alkylbenzenes is similar. Among C8H10 isomers, the reactivity of pure ethylbenzene and o-xylene and their blends with n-heptane showed a complex competing effect between the difference in CH bond energy and the existence of intermediate/low-temperature chemistry caused by adjacent methyl pairs. A similar structural impact was also observed for C9H12 isomers and their blends with n-heptane, while the influence of CH bond energy was more noticeable than C8H10 molecules. Kinetic simulations of the alkylbenzene/n-heptane blends highlighted the effect caused by adjacent methyl pairs that is referred to as the “ortho effect”. Analysis of ethylbenzene and o-xylene showed that o-xylene’s intermediate/low-temperature pathways initiated by benzylperoxy radical – benzylhydroperoxide isomerization (RO2 – QOOH) produce additional active radicals such as OH and CH2O, which accelerates the oxidation chemistry of more reactive n-heptane. This study provides knowledge on the blending effect of alkylbenzene compounds with n-heptane on their ignition characteristics that is useful to develop surrogates that can better mimic the reactivity of real fuels.
-
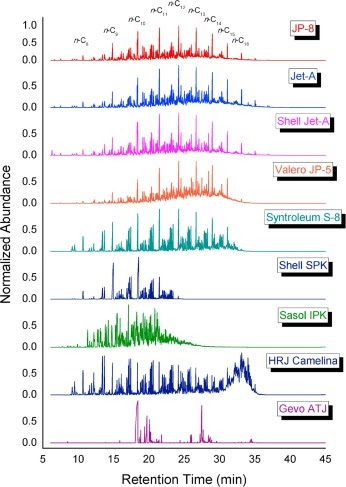 Experimental characterization of jet fuels under engine relevant conditions – Part 2: Insights on optimization approach for surrogate formulationDongil Kang, Doohyun Kim, Vickey Kalaskar, André Boehman, and Angela VioliFuel, Dec 2019
Experimental characterization of jet fuels under engine relevant conditions – Part 2: Insights on optimization approach for surrogate formulationDongil Kang, Doohyun Kim, Vickey Kalaskar, André Boehman, and Angela VioliFuel, Dec 2019Computational combustion modeling is an essential complementary tool to engine experiments and the combination of computational fluid dynamics and detailed chemical kinetics provides the promise for optimizing engine performance. However, predicting the effects of chemical and physical properties of fuels on engine performance is a great challenge since transportation fuels are composed of several hundreds to thousands of chemical species. Surrogates are simpler representations of real fuels and are comprised of selected species of known concentrations that exhibit combustion characteristics similar to those of the real fuels. In this paper, drawing from our previous work on surrogate formulations, we investigate the effects of surrogate components on the autoignition characteristics of conventional and alternative jet fuels, using a combination of experimental and modeling approaches. As target fuels, we analyzed a conventional jet fuel (Jet-A) and two alternative jet fuels (coal-derived IPK, natural-gas-derived S8). Experimental data on the properties of surrogate mixtures, such as liquid density and threshold sooting index, their combustion behaviors, together with those from their corresponding target real fuels are compared and analyzed using predictions obtained from the surrogate model. Results from a modified CFR engine show that the ignition reactivity of Jet-A and Sasol IPK surrogates are stronger than their target fuels, while the S8 surrogate displayed an ignition behavior very similar to the target S8. Results from a constant volume spray combustion chamber provided reasonable agreements between the surrogates and their target jet fuels in terms of physical and chemical ignition delay times and apparent heat release trends, with the S8 surrogate showing the best agreement. In addition, surrogate mixtures that were modified from the original Jet-A and IPK surrogates to achieve a better agreement with CFR experiments performed poorly when tested in a spray chamber. These results imply that the agreement between the behaviors of surrogates and real fuels in one device or in one condition does not guarantee similarity in other devices or in other conditions. This study also highlights the need for improvements in the current surrogate formulation methodologies to provide a more universal emulation of the autoignition behaviors of target transportation fuels.
-
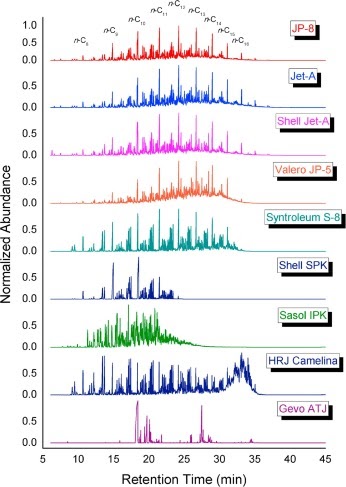 Experimental characterization of jet fuels under engine relevant conditions – Part 1: Effect of chemical composition on autoignition of conventional and alternative jet fuelsDongil Kang, Doohyun Kim, Vickey Kalaskar, Angela Violi, and André L. BoehmanFuel, Dec 2019
Experimental characterization of jet fuels under engine relevant conditions – Part 1: Effect of chemical composition on autoignition of conventional and alternative jet fuelsDongil Kang, Doohyun Kim, Vickey Kalaskar, Angela Violi, and André L. BoehmanFuel, Dec 2019Conventional and synthetic alternative jet fuels possess distinctive chemical compositions and physical/chemical properties since they are produced through different production processes. In this paper, the autoignition characteristics of four conventional aviation jet fuels (Jet-A POSF-4658, Jet-A POSF-10325, JP-5 POSF-10289, JP-8 POSF-6169), five alternative jet fuels (Syntroleum S-8 (S8), Shell synthetic paraffinic kerosene (Shell SPK), Sasol iso-paraffinic kerosene (Sasol IPK), hydro-processed renewable jet (HRJ8), alcohol to jet (ATJ), and 50/50 vol% blends of JP-8/alternative jet fuels were explored. The objective of the current work is to understand how the chemical composition of the fuels affects the gas phase chemical kinetic behavior and the ignition process of the liquid spray in an engine-relevant environment, thereby ultimately enabling their use in military ground vehicles with compression ignition engines. A modified Cooperative Fuel Research (CFR) motored engine was utilized and the gas phase ignition behavior was observed over a wide range of temperatures and pressures, while varying the compression ratio. In addition, an optically accessible constant volume spray combustion chamber was employed to observe significant differences in the global heat release characteristics and physical/chemical ignition delay times of synthetic alternative jet fuels in comparison to the conventional jet fuels. The results of this study show that synthetic alternative jet fuels with predominantly linear and lightly-branched alkane content (S8, Shell SPK and HRJ8) provide stronger low-temperature ignition characteristics, while other types of synthetic alternative jet fuels with high content of highly-branched alkanes (Sasol IPK and ATJ) exhibit weaker low-temperature ignition characteristics, when compared to conventional jet fuels (JP-8, Jet-A and JP-5). Their unique fundamental ignition behaviors, including the percentage of low-temperature heat release (% LTHR), the critical compression ratio (CCR), and the critical equivalence ratio (\ensuremathφcrit), are strongly related to the dominant chemical compositions of the fuel in a motored engine.
- Two-stage ignition behavior and octane sensitivity of toluene reference fuels as gasoline surrogateDoohyun Kim, Charles K. Westbrook, and Angela VioliCombustion and Flame, Dec 2019
Current approaches to improve the efficiency of Spark-Ignition (SI) gasoline engines have been focusing on turbocharging, increasing the compression ratio, and pursuing advanced low-temperature combustion concepts. In order to maximize these strategies, it is important to optimize the knock resistance of the fuel, and therefore knowledge of the sensitivity of the ignition process under a wide range of engine operating conditions is required. Octane sensitivity (OS), which is defined as the difference between Research Octane Number (RON) and Motored Octane Number (MON), has been introduced to represent how fuel’s ignition reactivity changes relative to the primary reference fuels (n-heptane/iso-octane) within RON/MON conditions. Previous works have indicated that OS is intimately related to low temperature reactivity of the fuel, which can be revealed as two-stage heat release characteristics during an ignition event. Prompted by these findings, in this paper, we investigate the relationship between two-stage ignition behavior and OS, using chemical kinetic simulations of 24 Toluene Reference Fuels (TRFs)/ethanol blends. TRFs are ternary mixtures of n-heptane/iso-octane/toluene, which is capable of capturing aromatic content and positive values of OS of real gasoline fuels. Simulation results show that fuels with weak or no two-stage ignition behavior tend to have high OS, due to their lack of Negative Temperature Coefficient (NTC) effect and high sensitivity in ignition delay time. Leveraging such observations, we develop a correlation between two-stage behavior and OS as an OS prediction method. Two metrics that represent the strength of the two-stage ignition behavior are proposed and used as OS predictors, which are Low Temperature Heat Release percentage (LTHR%) and Heat Release Rate at the end of first stage (HRRinf) calculated from a simple kinetic simulation. Regression analysis shows a clear trend between decreases in the proposed two-stage behavior metrics and increases in the value of OS of the fuel. We also test the new metric (LTHR%) using simulation results of 0-D reactors with imposed pressure time histories obtained from engine experiments, as well as using different TRF kinetic mechanisms. The results demonstrate the effectiveness of the metric as a representation of the two-stage ignition behavior in practical combustion systems, highlighting the importance of the proposed relationship, and its potential as a simple and effective OS predictor.
-
 Spatial dependence of the growth of polycyclic aromatic compounds in an ethylene counterflow flameQi Wang, Paolo Elvati, Doohyun Kim, K. Olof Johansson, Paul E. Schrader, and 2 more authorsCarbon, Dec 2019
Spatial dependence of the growth of polycyclic aromatic compounds in an ethylene counterflow flameQi Wang, Paolo Elvati, Doohyun Kim, K. Olof Johansson, Paul E. Schrader, and 2 more authorsCarbon, Dec 2019The complex environments that characterize combustion systems can influence the distribution of gas-phase species, the relative importance of various growth mechanisms and the chemical and physical characteristics of the soot precursors generated. In order to provide molecular insights on the effect of combustion environments on the formation of gas-phase species, in this paper, we study the temporal and spatial dependence of soot precursors growth mechanisms in an ethylene/oxygen/argon counterflow diffusion flame. As computational tools of investigation, we included fluid dynamics simulations and stochastic discrete modeling. Results show the relative importance of various reaction pathways in flame, with the hydrogen-abstraction-acetylene-addition mechanism contributing to the formation of pure hydrocarbons near the stagnation plane, and oxygen chemistry prevailing near the maximum temperature region, where the concentration of atomic oxygen reaches its peak and phenols, ethers and furan-embedded species are formed. The computational results show excellent agreement with measurements obtained using aerosol mass spectrometry coupled with vacuum-ultraviolet photoionization. Knowledge acquired in this study can be used to predict the type of compounds formed in various locations of the flame and eventually provide insights on the environmental parameters that influence the growth of soot precursors. Additionally, the results reported in this paper highlight the importance of modeling counterflow flames in two or three dimensions to capture the spatial dependence of growth mechanisms of soot precursors.
2018
-
 Homo-dimerization of oxygenated polycyclic aromatic hydrocarbons under flame conditionsP. Elvati and A. VioliFuel, Dec 2018
Homo-dimerization of oxygenated polycyclic aromatic hydrocarbons under flame conditionsP. Elvati and A. VioliFuel, Dec 2018Homogeneous nucleation is identified as one of the steps responsible for the formation of the critical nuclei that define the transition from gas-phase species to soot formation particles. However, the effect of molecular properties (like size, composition, and shape) on their propensity to physically bind, for a time long enough to initiate particle nucleation, has not been thoroughly investigated. In this work, we analyze the effect of oxygen content on the homodimerization propensity of different polycyclic aromatic compounds (PACs) using molecular dynamics simulations coupled with well-tempered Metadynamics enhanced sampling. We analyzed species in two mass groups, with a molecular weight ranging from 448 \ensuremathμ to 696 \ensuremathμ, and with varying oxygen content, mostly present as ethers. The reconstructed free energy profiles show that, besides mass, the presence of oxygenated groups influences the tendency of PACs to bind, with oxygenated compounds forming dimers that are less stable than the corresponding polycyclic aromatic hydrocarbons. Although this work represents only the first step towards a model for PACs dimerization that include oxygen, these results suggest that oxygenated compounds are less likely to condense onto soot particles.
-
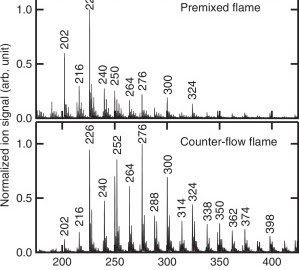 Hydrocarbons for the next generation of jet fuel surrogatesDoohyun Kim and Angela VioliFuel, Dec 2018
Hydrocarbons for the next generation of jet fuel surrogatesDoohyun Kim and Angela VioliFuel, Dec 2018Fuel surrogates are a critical component for the detailed combustion modeling of real transportation fuels. Indeed, the numerical study of engine combustion requires the coupling of computational fluid dynamics and chemical kinetic models, and therefore a limited number of chemical species and reactions can be employed due to current numerical capabilities. As a consequence, surrogates are adopted to simulate the behavior of real fuels. In this study, we evaluate various hydrocarbon molecules that can be employed as next generation surrogate components for conventional and alternative jet fuels. Species considered in this study have smaller number of kinetic data as compared to molecules that are currently used in jet fuel surrogates, but they possess greater physical relevance and the potential to achieve closer emulation of properties when used as jet fuel surrogate components. Using a surrogate optimizer model, we analyze various mixtures that can emulate a petroleum-derived jet fuel (Jet-A POSF-4658) and a coal-derived jet fuel (IPK POSF-5642). The results show that n-tetradecane and n-dodecane are suitable normal alkane representatives for jet fuels. Also, the use of three C9 alkylbenzenes (n-propyl-, 1,2,4-trimethyl-, 1,3,5-trimethyl-benzene) leads to surrogate mixtures with an aromatic content and a distillation curve that matches the experimental values of Jet-A much better than mixtures that contain toluene or C10 alkylbenzenes. In addition, the optimization results with three new branched alkanes for the target IPK show that 2,2,4,6,6-pentamethylheptane is a promising surrogate component for representing low ignition quality highly-branched alkanes in jet fuels. This study highlights the need for experimental studies and further kinetic model development for these molecules, which will benefit the surrogate development for the wide variety of jet fuels in the future.
2017
-
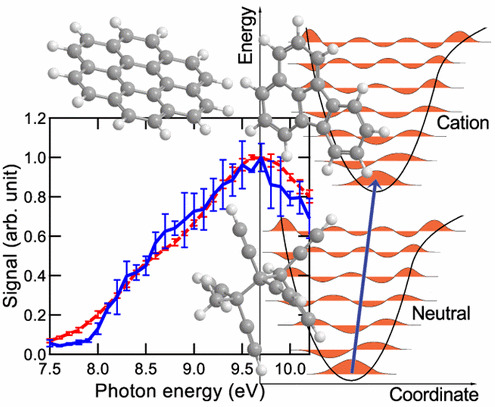 Critical Assessment of Photoionization Efficiency Measurements for Characterization of Soot-Precursor SpeciesK. Olof Johansson, Judit Zádor, Paolo Elvati, Matthew F. Campbell, Paul E. Schrader, and 4 more authorsThe Journal of Physical Chemistry A, Jun 2017
Critical Assessment of Photoionization Efficiency Measurements for Characterization of Soot-Precursor SpeciesK. Olof Johansson, Judit Zádor, Paolo Elvati, Matthew F. Campbell, Paul E. Schrader, and 4 more authorsThe Journal of Physical Chemistry A, Jun 2017We present a critical evaluation of photoionization efficiency (PIE) measurements coupled with aerosol mass spectrometry for the identification of condensed soot-precursor species extracted from a premixed atmospheric-pressure ethylene/oxygen/nitrogen flame. Definitive identification of isomers by any means is complicated by the large number of potential isomers at masses likely to comprise particles at flame temperatures. This problem is compounded using PIE measurements by the similarity in ionization energies and PIE-curve shapes among many of these isomers. Nevertheless, PIE analysis can provide important chemical information. For example, our PIE curves show that neither pyrene nor fluoranthene alone can describe the signal from C16H10 isomers and that coronene alone cannot describe the PIE signal from C24H12 species. A linear combination of the reference PIE curves for pyrene and fluoranthene yields good agreement with flame-PIE curves measured at 202 u, which is consistent with pyrene and fluoranthene being the two major C16H10 isomers in the flame samples, but does not provide definite proof. The suggested ratio between fluoranthene and pyrene depends on the sampling conditions. We calculated the values of the adiabatic-ionization energy (AIE) of 24 C16H10 isomers. Despite the small number of isomers considered, the calculations show that the differences in AIEs between several of the isomers can be smaller than the average thermal energy at room temperature. The calculations also show that PIE analysis can sometimes be used to separate hydrocarbon species into those that contain mainly aromatic rings and those that contain significant aliphatic content for species sizes investigated in this study. Our calculations suggest an inverse relationship between AIE and the number of aromatic rings. We have demonstrated that further characterization of precursors can be facilitated by measurements that test species volatility.
-
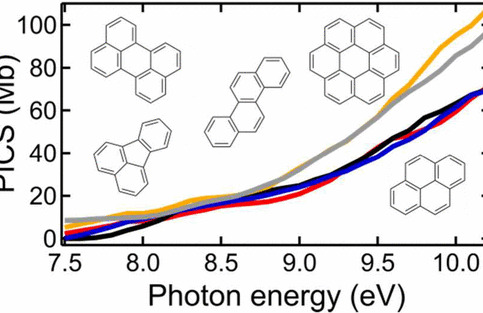 Photoionization Efficiencies of Five Polycyclic Aromatic HydrocarbonsK. Olof Johansson, Matthew F. Campbell, Paolo Elvati, Paul E. Schrader, Judit Zádor, and 4 more authorsThe Journal of Physical Chemistry A, Jun 2017
Photoionization Efficiencies of Five Polycyclic Aromatic HydrocarbonsK. Olof Johansson, Matthew F. Campbell, Paolo Elvati, Paul E. Schrader, Judit Zádor, and 4 more authorsThe Journal of Physical Chemistry A, Jun 2017We have measured photoionization-efficiency curves for pyrene, fluoranthene, chrysene, perylene, and coronene in the photon energy range of 7.5–10.2 eV and derived their photoionization cross-section curves in this energy range. All measurements were performed using tunable vacuum ultraviolet (VUV) radiation generated at the Advanced Light Source synchrotron at Lawrence Berkeley National Laboratory. The VUV radiation was used for photoionization, and detection was performed using a time-of-flight mass spectrometer. We measured the photoionization efficiency of 2,5-dimethylfuran simultaneously with those of pyrene, fluoranthene, chrysene, perylene, and coronene to obtain references of the photon flux during each measurement from the known photoionization cross-section curve of 2,5-dimethylfuran.
- The Effects of Injection Timing and Injected Fuel Mass on Local Charge Conditions and Emissions for Gasoline Direct Injection EnginesDoohyun Kim, Angela Violi, and André BoehmanOct 2017
Increased Particulate Matter (PM) emissions from Gasoline Direct Injection (GDI) engines compared to conventional Port Fuel Injection (PFI) engines have been raising concerns because of the PM’s detrimental health effects and the stringent emissions regulations. One of the widely accepted hypotheses is that local rich pockets inside the combustion chamber are the primary reason for the increased PM emissions. In this paper, we investigate the effects of injection strategies on the charge composition and local thermodynamic conditions of a light duty GDI engine, and determine their impact on PM emissions. The operation of a 1.6L GDI engine is simulated using a 3-D Computational Fluid Dynamics (CFD) code. Combustion characteristics of a 3-component gasoline surrogate (n-heptane/iso-octane/toluene) are analyzed and the effects of injection timing (300\textdegree vs 240\textdegree vs 180\textdegree BTDC) and injected fuel mass (globally stoichiometric vs fuel rich) are explored at 2000 rpm, 9.5 bar BMEP condition, focusing on the homogeneity of the charge and the formation of the gaseous species that are soot precursors. The results indicate that when the physical time for air/fuel mixing is not long enough, fuel-rich pockets are present until combustion occurs, where high concentrations of soot precursors are found, such as acetylene and pyrene. In addition, simulation results indicate that the location of wetted surface as well as the in-cylinder flow structure induced by the fuel jet hitting the piston bowl is significantly influenced by varying the injection timing, which affects subsequent air/fuel mixing. When the injected fuel mass is increased, the equivalence ratio distribution inside the combustion chamber shifts toward fuel-rich side, generating more mixtures with \ensuremathΦ \\> 1.5, where formation of acetylene and pyrene are favored.
-
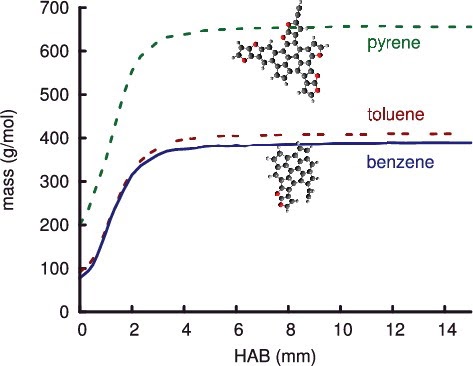 The effect of reaction mechanisms on the formation of soot precursors in flamesTyler Dillstrom and Angela VioliCombustion Theory and Modelling, Oct 2017
The effect of reaction mechanisms on the formation of soot precursors in flamesTyler Dillstrom and Angela VioliCombustion Theory and Modelling, Oct 2017Nanoparticles formed in gas-phase combustion environments are ubiquitous in modern society as hazardous pollutants. Due to the deleterious health effects of carbonaceous nanoparticles it is important to gain a better understanding of the fundamental processes that lead to their formation and growth. We have utilised our Stochastic NAnoParticle Simulator software to model the growth of soot precursor species in a laminar premixed benzene–air flame. We implemented a new set of pathways containing oxygenation reactions and assessed the effects on the growth mechanisms. The inclusion of oxygenation growth pathways with the previously established hydrocarbon growth pathways creates a much larger set of potential configurations for the particle to explore and thus the rate of growth is faster. Hydrogen abstractions and acetylene additions are still the most common events the particle experiences during its evolution trajectory; however, a significant amount of ethers and oxygen-containing rings are formed. Though the role of soot precursor oxygenation is not completely solved, it certainly adds layers of complexity to the nanoparticles’ formation process and likely influences the formation of soot particles.
-
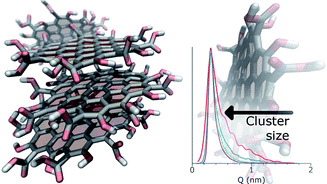 Graphene quantum dots: effect of size, composition and curvature on their assemblyPaolo Elvati, Elizabeth Baumeister, and Angela VioliRSC Adv., Oct 2017
Graphene quantum dots: effect of size, composition and curvature on their assemblyPaolo Elvati, Elizabeth Baumeister, and Angela VioliRSC Adv., Oct 2017Graphene Quantum Dots (GQDs) are a relatively new class of molecules that have ignited tremendous research interest due to their extraordinary and tunable optical, electrical, chemical and structural properties. In this work, we report a molecular-level elucidation of the key mechanisms and physical–chemical factors controlling the assembly and stability of nanostructures formed by GQDs in an aqueous environment, using molecular dynamics simulations. We observe the general tendency to form small aggregates and three recurring configurations, one of them with a single layer of water separating two GQDs. The type and characteristics of the structure are mostly determined by the hydrophobicity of the GQDs as well as the steric hindrance of the dangling groups. The composition of the terminal groups plays a key role in determining the configuration of the GQDs, which is also markedly affected by the formation of clusters. Notably, the aggregated GQDs assume strongly correlated shapes and, in some cases, display a radically different conformation distribution compared to single molecules. This cooperative effect prolongs the lifetime of the GQD configurations and can explain the observed persistence of chiral conformations that are only marginally more stable than their specular images.
- Oxygen driven soot formationP. Elvati, V.t. Dillstrom, and A. VioliProceedings of the Combustion Institute, Oct 2017
The emission standards of combustion have been steadily reduced in recent years, and a large research effort has been focused on lowering the emissions of hydrocarbons and particulate matter. Addition of oxygenates to fuel reduces these pollutants. In this paper, we report on a detailed investigation of the growth mechanisms for gas-phase species in ethylene/air and ethylene/ethanol/air flames in order to assess the importance of various chemical mechanisms for the molecular growth of soot precursors. We employ a variety of computational techniques that include stochastic and deterministic methods to study the formation of gas-phase species and their growth into soot precursors via chemical and physical mechanisms. We have drawn the following conclusions: 1) the chemistry of oxygenated compounds (specifically the high concentrations of O2 and OH) is critical to reproduce the experiments; 2) using free energy simulations, we found out that the tendency of molecules to form dimers is mainly affected by the molecular shape rather than the mass of the aggregate. Finally we propose a different mechanism for the growth of soot precursors based on radical–radical recombination to form molecules of high molecular masses (>1200u). These structures are then likely to promote physical aggregation to further the growth mechanism.
- A Data-Driven Sparse-Learning Approach to Model Reduction in Chemical Reaction NetworksFarshad Harirchi, Omar A. Khalil, Sijia Liu, Paolo Elvati, Angela Violi, and 1 more authorOct 2017
In this paper, we propose an optimization-based sparse learning approach to identify the set of most influential reactions in a chemical reaction network. This reduced set of reactions is then employed to construct a reduced chemical reaction mechanism, which is relevant to chemical interaction network modeling. The problem of identifying influential reactions is first formulated as a mixed-integer quadratic program, and then a relaxation method is leveraged to reduce the computational complexity of our approach. Qualitative and quantitative validation of the sparse encoding approach demonstrates that the model captures important network structural properties with moderate computational load.
-
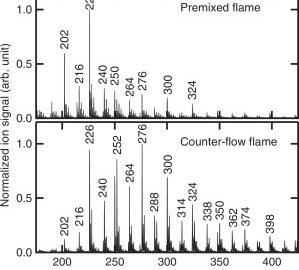 Radical-radical reactions, pyrene nucleation, and incipient soot formation in combustionK. Olof Johansson, Tyler Dillstrom, Paolo Elvati, Matthew F. Campbell, Paul E. Schrader, and 5 more authorsProceedings of the Combustion Institute, Oct 2017
Radical-radical reactions, pyrene nucleation, and incipient soot formation in combustionK. Olof Johansson, Tyler Dillstrom, Paolo Elvati, Matthew F. Campbell, Paul E. Schrader, and 5 more authorsProceedings of the Combustion Institute, Oct 2017We present a combined experimental and probabilistic simulation study of soot-precursor. The experiments were conducted using aerosol mass spectrometry coupled with tunable vacuum ultraviolet radiation from the Advanced Light Source at Lawrence Berkeley National Laboratory. Mass spectra and photoionization efficiency (PIE) curves of soot precursor species were measured at different heights in a premixed flat flame and in a counter-flow diffusion flame fueled by ethylene and oxygen. The PIE curves at the pyrene mass from these flames were compared with reference PIE scans recorded for pyrene. The results demonstrate that other C16H10 isomers than pyrene are major components among species condensed onto incipient soot in this study, which is in agreement with the simulations. Species with mass 202u only have a high prevalence in incipient soot particles drawn from the premixed flame, but hydrocarbon species with sizes in the range 200–400u are important to incipient-soot formation in both flames. The simulations predict that some species form through combination reactions involving relatively large radicals and bypass traditional molecular-growth pathways through addition of small hydrocarbon species. The experimental results support this prediction; they demonstrate that these species have higher relative abundances in particles formed close to the fuel outlet than smaller, lighter molecular species and indicate that these species are important to early formation of incipient-soot precursors. The results also imply that a leading role in incipient-soot precursor formation is played by species with lower thermal stability than the even-carbon numbered, unsubstituted polycyclic aromatic hydrocarbons known as “stabilomers”.
- Experimental Validation of Jet Fuel Surrogates in an Optical EngineT Kim, X Luo, M Al-Sadoon, MC Lai, M Jansons, and 4 more authorsIn , Oct 2017
Three jet fuel surrogates were compared against their target fuels in a compression ignited optical engine under a range of start-of-injection temperatures and densities. The jet fuel surrogates are representative of petroleum-based Jet-A POSF-4658, natural gas-derived S-8 POSF-4734 and coal-derived Sasol IPK POSF-5642, and were prepared from a palette of n-dodecane, n-decane, decalin, toluene, iso-octane and iso-cetane. Optical chemiluminescence and liquid penetration length measurements as well as cylinder pressure-based combustion analyses were applied to examine fuel behavior during the injection and combustion process. HCHO_∗ emissions obtained from broadband UV imaging were used as a marker for low temperature reactivity, while 309 nm narrow band filtered imaging was applied to identify the occurrence of OH_∗, autoignition and high temperature reactivity. Jet-A and S-8 were well represented by their surrogate fuels under the conditions examined, while the Sasol-IPK surrogate emulated the low temperature heat release but had a shorter high temperature ignition delay compared to the target fuel. The IPK surrogate was also found to have a slightly greater liquid penetration length and greater low-temperature reactivity.
-
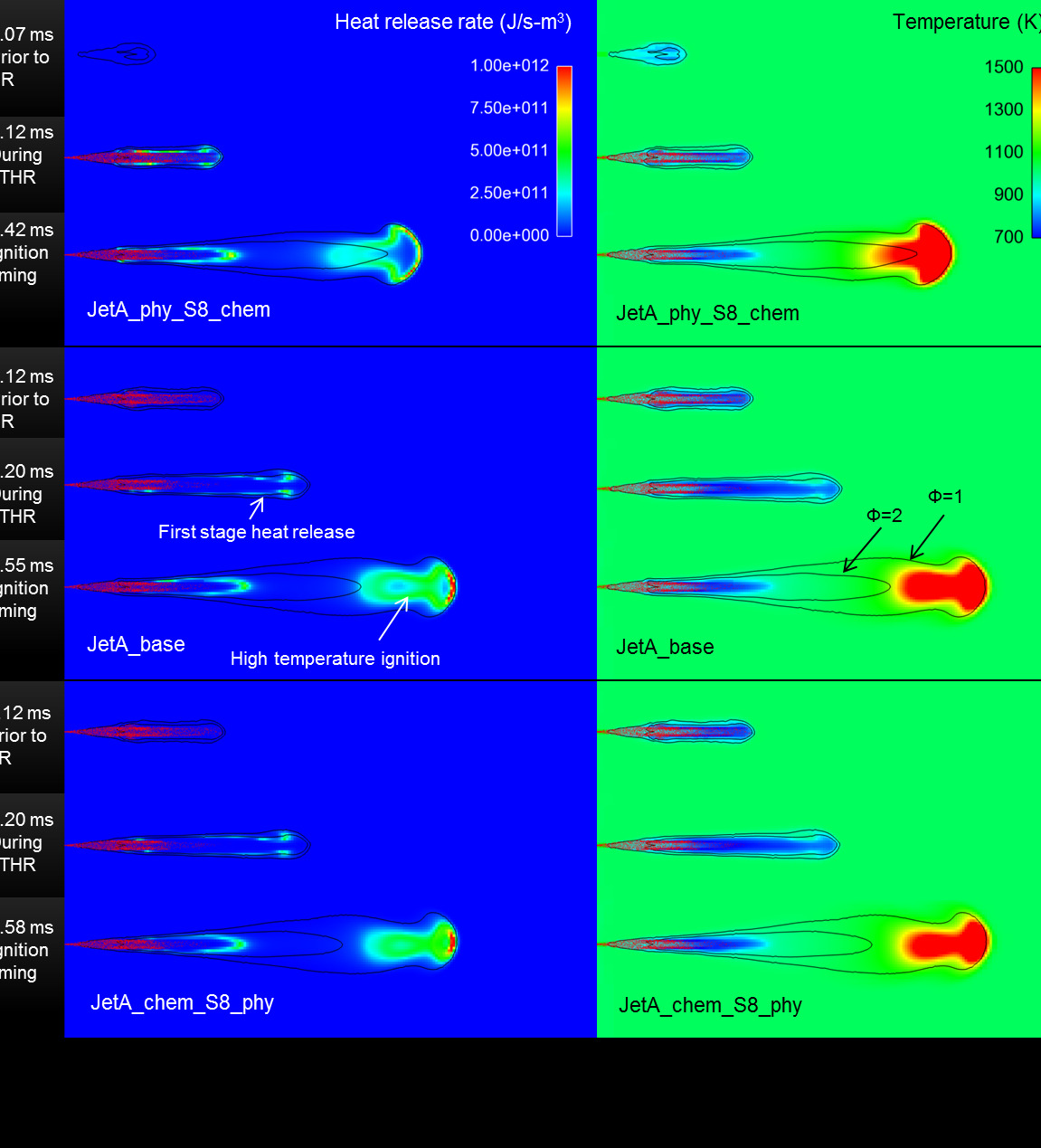 The Relative Importance of Fuel Oxidation Chemistry and Physical Properties to Spray IgnitionDoohyun Kim, J Martz, and Angela VioliOct 2017
The Relative Importance of Fuel Oxidation Chemistry and Physical Properties to Spray IgnitionDoohyun Kim, J Martz, and Angela VioliOct 2017The ignition delay time for direct injection compression ignition engines is determined by complex physical and chemical phenomena that prepare the injected liquid fuel for gas phase ignition. In this work, Computational Fluid Dynamics (CFD) simulations of a reacting spray within a constant volume spray chamber are conducted to investigate the relative importance of liquid fuel physical properties and oxidation chemistry on the ignition delay time. The simulations use multi-component surrogates that emulate the physical and chemical properties of petroleum-derived (Jet-A) and natural-gas-derived (S-8) jet fuels. Results from numerical experiments isolating the fuel physical property and chemistry effects show that fuel chemistry is significantly more important to ignition delay than fuel physical properties under the conditions studied. In addition, as the air charge temperature increases, the effects of physical properties and oxidation chemistry decrease, indicating that the effect of variation in fuel properties on ignition timing may be mitigated through increased pre-ignition charge temperatures.
- A six-component surrogate for emulating the physical and chemical characteristics of conventional and alternative jet fuels and their blendsDoohyun Kim, Jason Martz, Andrew Abdul-nour, Xin Yu, Marcis Jansons, and 1 more authorCombustion and Flame, Oct 2017
Conventional and alternative jet fuels, such as petroleum-derived Jet-A, coal-derived IPK, and natural-gas-derived S-8, display significant chemical and physical fuel property differences that influence their ignition characteristics. The current work addresses the need for surrogate mixtures capable of emulating the various properties of these fuels and their select blends, which are often used within compression ignited engines for acceptable ignition behavior. A six-component surrogate palette is proposed with species that are readily available within recent kinetic mechanisms, including n-dodecane, n-decane, iso-cetane, iso-octane, decalin, and toluene. The use of these species allows for a seamless compositional transition between the neat target jet fuels and their blends. The surrogate optimizer, which includes various correlations and models to estimate properties of model mixtures, is used to determine the surrogate composition that best matches target fuel properties. For an accurate ignition quality prediction during the optimization, a non-linear Derived Cetane Number regression equation is generated from Ignition Quality Tester experiments of 76 surrogate component mixtures. The newly formulated surrogates and their blends successfully capture the wide range of properties present within the target fuels, including temperature-dependent physical properties such as density, viscosity, specific heat, and volatility, along with experimental ignition delays obtained from a constant volume spray chamber. Kinetic modeling with a detailed mechanism showed that predicted ignition delay times are in good agreement with shock tube and rapid compression machine ignition delay experiments. A sensitivity analysis with variations in the composition of the Jet-A surrogate showed that its calculated ignition delay times are most sensitive to the composition of n-dodecane among the four Jet-A surrogate constituents over the range of temperatures and pressures examined.
2016
-
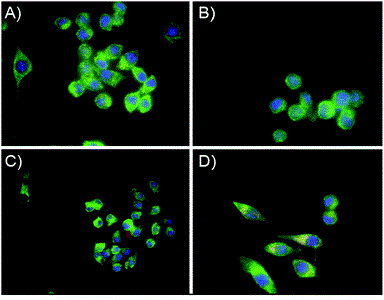 C60 fullerene localization and membrane interactions in RAW 264.7 immortalized mouse macrophagesK A Russ, P Elvati, T L Parsonage, A Dews, J A Jarvis, and 6 more authorsNanoscale, Feb 2016
C60 fullerene localization and membrane interactions in RAW 264.7 immortalized mouse macrophagesK A Russ, P Elvati, T L Parsonage, A Dews, J A Jarvis, and 6 more authorsNanoscale, Feb 2016There continues to be a significant increase in the number and complexity of hydrophobic nanomaterials that are engineered for a variety of commercial purposes making human exposure a significant health concern. This study uses a combination of biophysical, biochemical and computational methods to probe potential mechanisms for uptake of C60 nanoparticles into various compartments of living immune cells. Cultures of RAW 264.7 immortalized murine macrophage were used as a canonical model of immune-competent cells that are likely to provide the first line of defense following inhalation. Modes of entry studied were endocytosis/pinocytosis and passive permeation of cellular membranes. The evidence suggests marginal uptake of C60 clusters is achieved through endocytosis/pinocytosis, and that passive diffusion into membranes provides a significant source of biologically-available nanomaterial. Computational modeling of both a single molecule and a small cluster of fullerenes predicts that low concentrations of fullerenes enter the membrane individually and produce limited perturbation; however, at higher concentrations the clusters in the membrane causes deformation of the membrane. These findings are bolstered by nuclear magnetic resonance (NMR) of model membranes that reveal deformation of the cell membrane upon exposure to high concentrations of fullerenes. The atomistic and NMR models fail to explain escape of the particle out of biological membranes, but are limited to idealized systems that do not completely recapitulate the complexity of cell membranes. The surprising contribution of passive modes of cellular entry provides new avenues for toxicological research that go beyond the pharmacological inhibition of bulk transport systems such as pinocytosis.
-
 Chiral Graphene Quantum DotsNozomu Suzuki, Yichun Wang, Paolo Elvati, Zhi-Bei Qu, Kyoungwon Kim, and 8 more authorsACS Nano, Feb 2016
Chiral Graphene Quantum DotsNozomu Suzuki, Yichun Wang, Paolo Elvati, Zhi-Bei Qu, Kyoungwon Kim, and 8 more authorsACS Nano, Feb 2016Chiral nanostructures from metals and semiconductors attract wide interest as components for polarization-enabled optoelectronic devices. Similarly to other fields of nanotechnology, graphene-based materials can greatly enrich physical and chemical phenomena associated with optical and electronic properties of chiral nanostructures and facilitate their applications in biology as well as other areas. Here, we report that covalent attachment of l/d-cysteine moieties to the edges of graphene quantum dots (GQDs) leads to their helical buckling due to chiral interactions at the “crowded” edges. Circular dichroism (CD) spectra of the GQDs revealed bands at ca. 210–220 and 250–265 nm that changed their signs for different chirality of the cysteine edge ligands. The high-energy chiroptical peaks at 210–220 nm correspond to the hybridized molecular orbitals involving the chiral center of amino acids and atoms of graphene edges. Diverse experimental and modeling data, including density functional theory calculations of CD spectra with probabilistic distribution of GQD isomers, indicate that the band at 250–265 nm originates from the three-dimensional twisting of the graphene sheet and can be attributed to the chiral excitonic transitions. The positive and negative low-energy CD bands correspond to the left and right helicity of GQDs, respectively. Exposure of liver HepG2 cells to l/d-GQDs reveals their general biocompatibility and a noticeable difference in the toxicity of the stereoisomers. Molecular dynamics simulations demonstrated that d-GQDs have a stronger tendency to accumulate within the cellular membrane than l-GQDs. Emergence of nanoscale chirality in GQDs decorated with biomolecules is expected to be a general stereochemical phenomenon for flexible sheets of nanomaterials.
-
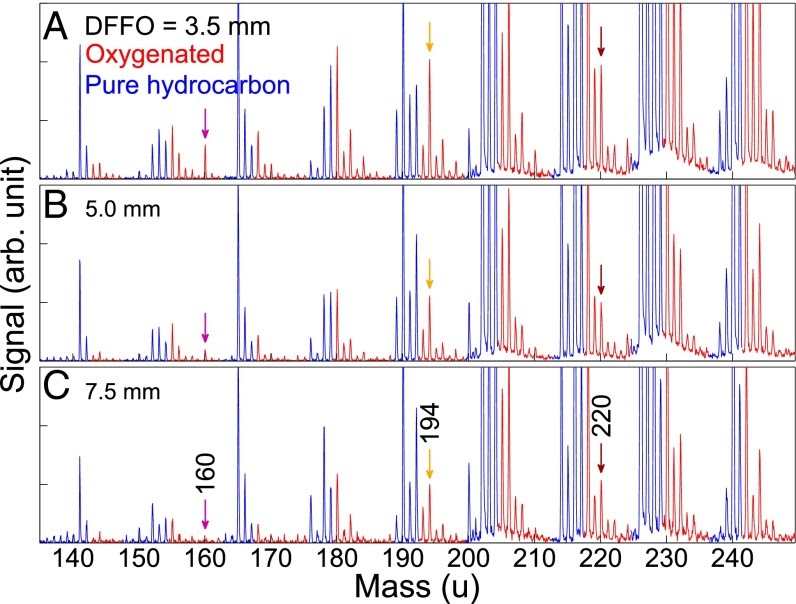 Formation and emission of large furans and oxygenated hydrocarbons from flamesK. Olof Johansson, Tyler Dillstrom, Matteo Monti, Farid El Gabaly, Matthew F. Campbell, and 6 more authorsProceedings of the National Academy of Sciences, Feb 2016
Formation and emission of large furans and oxygenated hydrocarbons from flamesK. Olof Johansson, Tyler Dillstrom, Matteo Monti, Farid El Gabaly, Matthew F. Campbell, and 6 more authorsProceedings of the National Academy of Sciences, Feb 2016Many oxygenated hydrocarbon species formed during combustion, such as furans, are highly toxic and detrimental to human health and the environment. These species may also increase the hygroscopicity of soot and strongly influence the effects of soot on regional and global climate. However, large furans and associated oxygenated species have not previously been observed in flames, and their formation mechanism and interplay with polycyclic aromatic hydrocarbons (PAHs) are poorly understood. We report on a synergistic computational and experimental effort that elucidates the formation of oxygen-embedded compounds, such as furans and other oxygenated hydrocarbons, during the combustion of hydrocarbon fuels. We used ab initio and probabilistic computational techniques to identify low-barrier reaction mechanisms for the formation of large furans and other oxygenated hydrocarbons. We used vacuum-UV photoionization aerosol mass spectrometry and X-ray photoelectron spectroscopy to confirm these predictions. We show that furans are produced in the high-temperature regions of hydrocarbon flames, where they remarkably survive and become the main functional group of oxygenates that incorporate into incipient soot. In controlled flame studies, we discovered ∼100 oxygenated species previously unaccounted for. We found that large alcohols and enols act as precursors to furans, leading to incorporation of oxygen into the carbon skeletons of PAHs. Our results depart dramatically from the crude chemistry of carbon- and oxygen-containing molecules previously considered in hydrocarbon formation and oxidation models and spearhead the emerging understanding of the oxidation chemistry that is critical, for example, to control emissions of toxic and carcinogenic combustion by-products, which also greatly affect global warming.
-
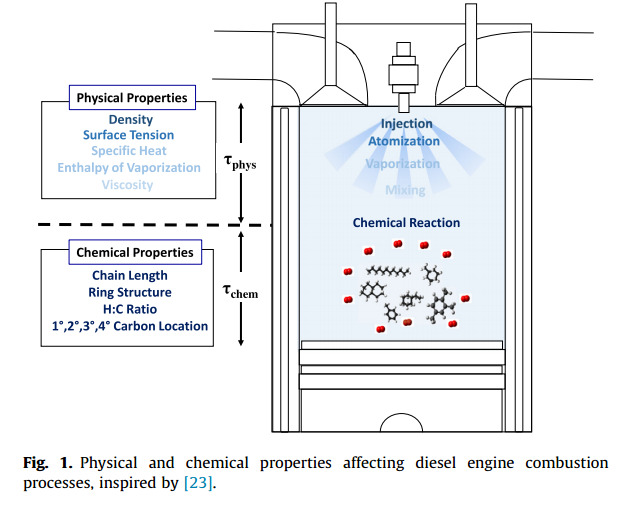 Experimental study of autoignition characteristics of Jet-A surrogates and their validation in a motored engine and a constant-volume combustion chamberDongil Kang, Vickey Kalaskar, Doohyun Kim, Jason Martz, Angela Violi, and 1 more authorFuel, Feb 2016
Experimental study of autoignition characteristics of Jet-A surrogates and their validation in a motored engine and a constant-volume combustion chamberDongil Kang, Vickey Kalaskar, Doohyun Kim, Jason Martz, Angela Violi, and 1 more authorFuel, Feb 2016The current study presents an experimental validation of jet aviation fuel surrogates formulated using a surrogate model-optimizer. Two surrogate fuel mixtures are used to emulate a practical Jet-A (POSF 4658) considering a series of physical and chemical processes in diesel engines. The surrogate mixtures consist of: n-dodecane/isocetane/methylcyclohexane (MCH)/toluene 0.3844/0.1484/0.2336/0.2336 on a molar basis (referred to as UM I), and n-dodecane/isocetane/decalin/toluene 0.2897/0.1424/0.3188/0.2491 on a molar basis (referred to as UM II). In the present study, a modified Cooperative Fuels Research (CFR) Octane Rating engine and an optically accessible, constant-volume spray combustion chamber are employed to investigate how the chemical and physical properties of these jet fuel surrogate mixtures affect the fundamental ignition behavior as compared to the targeted full boiling range Jet-A fuel (POSF 4658). The observations of ignition behavior include critical compression ratio and critical equivalence ratio, and % low temperature heat release, which are assessed using the motored engine (CFR), while physical and chemical ignition delays are measured using a modified Cetane Rating (CID 510) instrument under a wide range of air temperatures and oxygen dilution levels. The measured derived cetane number (DCN) for UM I shows a reasonably good match with the practical Jet-A fuel (POSF 4658), while gas-phase oxidation behavior is less, well-matched in the modified CFR engine. In contrast, although the measured DCN of UM II revealed a lower value than the estimated value using a surrogate optimizer, the ignition behavior is relatively well match with that of the target Jet-A in the modified CFR engine. The similarities in physical and chemical ignition delays as compared with the target Jet-A support the effectiveness of the UM II surrogate, while in contrast, the UM I surrogate only matched the chemical ignition delay. This is attributed to the importance of the surrogate component decalin, which improved the physical properties as compared to MCH, while the other three components were the same for the two surrogates.
-
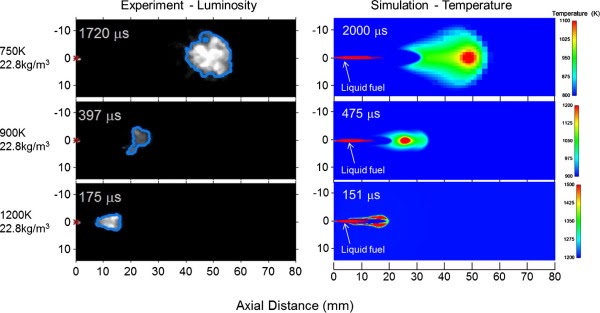 Effects of fuel physical properties on direct injection spray and ignition behaviorDoohyun Kim, Jason Martz, and Angela VioliFuel, Feb 2016
Effects of fuel physical properties on direct injection spray and ignition behaviorDoohyun Kim, Jason Martz, and Angela VioliFuel, Feb 2016CFD simulations of reacting fuel sprays were conducted to identify temperature-dependent physical properties of the liquid fuel that should be emulated by diesel and jet fuel surrogates during compression ignited combustion. Using a validated CFD model for an n-dodecane spray under diesel-relevant conditions, six physical properties of the liquid phase fuel (density, vapor pressure, viscosity, surface tension, heat of vaporization, and specific heat capacity) were perturbed covering the minimum and maximum property range of hydrocarbons widely used in recent diesel and jet fuel surrogates. Liquid fuel density, viscosity, vapor pressure, and specific heat had significant impact on liquid penetration length, causing a 4–16% change from the baseline n-dodecane case. The changes resulted from the liquid fuel’s influence on various physical phenomena, including droplet breakup, air entrainment and evaporation. For ignition delay, specific heat and density effects were most significant, with up to 10% changes from the baseline case. Specific heat perturbations affected the thermal energy necessary for fuel vaporization, hence local temperature development and mixture reactivity. Liquid density influenced the velocity of the fuel injection event, which modified turbulent mixing rates, low temperature heat release characteristics and the transition to high temperature ignition. The results of this study indicate that liquid density, specific heat, viscosity, and vapor pressure should be considered for surrogate development to properly capture the liquid penetration and ignition delay characteristics of the target fuel.
-
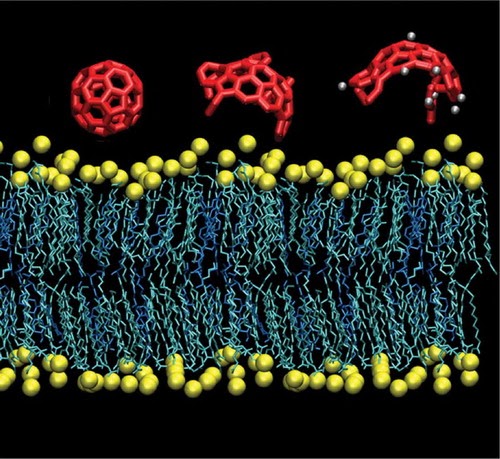 Effects of Combustion-Generated Nanoparticles on Cellular MembranesAngela VioliCombustion Science and Technology, Feb 2016
Effects of Combustion-Generated Nanoparticles on Cellular MembranesAngela VioliCombustion Science and Technology, Feb 2016In the environment, the process of combustion is the dominant pathway through which mankind continuously injects particles into the atmosphere at the present time. The most direct and serious risk related to these emissions is the direct absorption of these particles into the living systems of humans and animals through the process of respiration, especially in more urban environments. Little is known about the potential of low-level exposures to alter key biophysical functions, such as membrane form and function. This study employs molecular dynamics simulations to reconstruct the free energy landscapes of the mechanisms of entry of nanoparticles into biological cells. Specifically, we investigate the behavior of two nanoparticles produced in combustion conditions with different amphiphilic properties to assess the effect of chemical composition on the interactions with cellular membranes. Free energy calculations of nanoparticles interacting with a lipid bilayer composed of dimyristoylphosphatidylcholine and cholesterol show that the presence of hydroxyl groups on the nanoparticle makes the region where the lipids’ heads are solvated by water the most favorable position for these species. The hydrophobic nanoparticle shows a strong affinity for the center of the membrane. This study shows the importance of surface composition and suggests different mechanisms of interactions of nanoparticles with cellular membranes.
2015
-
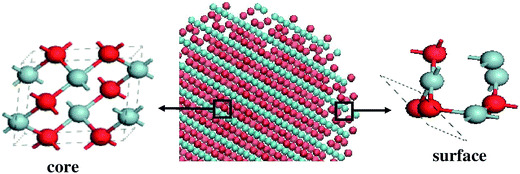 Size-and phase-dependent structure of copper(ii) oxide nanoparticlesAlauddin Ahmed, Paolo Elvati, and Angela VioliRSC Adv., Feb 2015
Size-and phase-dependent structure of copper(ii) oxide nanoparticlesAlauddin Ahmed, Paolo Elvati, and Angela VioliRSC Adv., Feb 2015Copper(ii) oxide (CuO) nanoparticles (NPs) have found numerous applications in electronics, optics, catalysis, energy storage, health, and water purification. Controlled synthesis of CuO NPs requires information on their nanoscale structure, which is expected to vary depending on the size, shape, phase, and most importantly, on the surface morphology. In this work, we report a detailed analysis of the structure of solid and melted CuO nanoparticles as functions of size and temperature at global and local scales, using molecular dynamics simulations. Comparisons of simulated X-ray diffraction profiles, mean bond lengths, average coordination numbers, and melting points with available experimental data support the modeling results. Melting points of CuO NPs vary linearly with the reciprocal of the diameters of NPs. The long-range order seen in solid nanoparticles with diameters greater than 6 nm gradually vanishes as size decreases, indicating the loss of translational symmetry of the lattice structure and the emergence of amorphous-like structure even below the melting point. Melted nanoparticles show liquid-like characteristics with only a short-range order. Mean bond lengths and average Cu–O coordination numbers of both solid and melted NPs indicate weakening of the structural stabilization for smaller NPs that leads to an increased deformation in the local atomic arrangement because of the lack of long-range interactions. For the cases studied, most of the structural features are independent of temperature, with the notable exception of the number of oxygen atoms coordinated to Cu. This latter quantity is indeed indicative of melting phase transition and can be used to compute the melting point accurately. Atoms on the surface of solid NPs show amorphous-like behavior even at temperatures well below the melting point of the NPs due to the limited coordination environment. This study represents a useful step towards the establishment of a structure–property relationship for CuO nanoparticles.
- Soot precursor formation and limitations of the stabilomer gridK.O. Johansson, J.Y.W. Lai, S.A. Skeen, D.M. Popolan-Vaida, K.R. Wilson, and 3 more authorsProceedings of the Combustion Institute, Feb 2015
We have combined experimental and theoretical approaches to gain new insight into the mechanisms of PAH growth and soot formation. The experimental approach involves aerosol-mass spectrometry in conjunction with vacuum-ultraviolet photoionization of volatile species vaporizing from particles sampled from an Ar-diluted C2H2/O2 counter-flow diffusion flame at nearly atmospheric pressure (700Torr). We recorded aerosol mass spectra at different distances from the fuel outlet for fixed ionization energies and in a fixed position while tuning the photoionization energy. The mass spectra contain a large distribution of peaks, highlighting the importance of small building blocks and showing a variety of chemical species that extends beyond the traditional classification of PAHs based on thermodynamic stability. In addition, we performed stochastic simulations of PAH growth in the flame in order to provide better insight into the chemical composition of species associated with peaks in the measured mass spectra. These simulations were conducted using a stochastic nanoparticle simulator (SNAPS). Synthesis of experimental and simulated results showed that peaks in the observed mass spectra generally consisted of a mixture of PAH isomers. At m/z=154 and 202, for example, experiments and simulations suggested that additional isomers than biphenyl and pyrene are important. Furthermore, the results highlight the importance of odd-carbon numbered species and complex growth paths. The experimental results suggest that species of higher masses can build up concentration ahead of species of lower masses. Our experimental results show, for example, that the peak at m/z=278 appears closer to the burner outlet than the peak at m/z=202, i.e., suggesting that a single monotonic growth mechanism is not enough.
- Towards a predictive model for polycyclic aromatic hydrocarbon dimerization propensityJeffrey S. Lowe, Jason Y.W. Lai, Paolo Elvati, and Angela VioliProceedings of the Combustion Institute, Feb 2015
Soot particles are a significant pollutant formed as the result of incomplete combustion. Particle nucleation significantly impacts the formation and morphology of soot particles, yet remains a key knowledge gap. To elucidate the process of nucleation, we have investigated the thermodynamic stability of dimers of polycyclic aromatic hydrocarbons (PAHs), towards developing a more comprehensive model for PAH clustering behavior. Using a computational methodology based on molecular dynamics and well-tempered Metadynamics, we quantified the impact of morphological parameters on homo-molecular dimerization, as well as the relative size of monomers on the stability of hetero-molecular dimers. The results illustrated the substantial impact of PAH mass and geometry on the stability of homo-molecular and hetero-molecular dimers at flame temperatures. In particular, dimer stability was found to depend most strongly on monomer mass, followed by solvent-accessible surface area. Additionally, hetero-molecular dimer stability was found to be largely determined by the size of the smallest monomer. Identifying relationships between PAH morphology and thermodynamic stability is a significant step towards a more comprehensive understanding of the physical interactions between PAHs. Altogether, this work presents a framework for elucidating the clustering behavior of arbitrary PAHs and will greatly impact understanding and modeling of particle nucleation and growth.
- Effects of a JP-8 surrogate and its components on soot in laminar, N2-diluted ethylene co-flow diffusion flames from 1 to 5atmAnne Geraldine Mouis, Thomas A. Litzinger, Yefu Wang, Venkatesh Iyer, Suresh Iyer, and 3 more authorsCombustion and Flame, Feb 2015
Experimental results are presented for changes in soot volume fraction resulting from the addition of a JP-8 surrogate and each of its components to a N2-diluted, C2H4 co-flow diffusion flame. The surrogate, which consists of 77% n-dodecane and 23% m-xylene by volume, was designed to match the threshold soot index of a nominal JP-8 fuel. Total carbon flow rate was constant for all experiments; pre-vaporized liquid fuel was added at two different levels: 2.5% and 5% of the total carbon flow rate. Tests were conducted at pressures from 1 to 5atm. The use of relatively small amounts of carbon from the liquid fuel resulted in a linear relationship between the peak soot volume fraction and the amount of carbon from the liquid fuel. In these experiments, the peak soot volume fraction was found to be vary with pressure according to a power-law relationship, consistent with prior work on pressure effects on soot. The surrogate fuel showed very similar trends to the JP-8, but yielded lower soot volume fractions. Simulation results for the flames with m-xylene capture the trends of increasing soot volume fraction with increasing carbon from the liquid fuel and with increasing pressure. However, the simulations show smaller increases than were observed in the experiments.
- A Fuel Surrogate Validation Approach Using a JP-8 Fueled Optically Accessible Compression Ignition EngineX Yu, X Luo, M Jansons, Doohyun Kim, J Martz, and 1 more authorFeb 2015
An experimental fuel surrogate validation approach is proposed for a compression ignition application, and applied to validate a Jet-A POSF 4658 fuel surrogate. The approach examines the agreement of both physical and chemical properties of surrogate and target fuels during validation within a real compression-ignition engine environment during four sequential but distinct combustion phases. In-cylinder Mie Scattering measurements are applied to evaporating sprays to compare the behavior of the surrogate, its target fuel, and for reference, n-heptane. Early mixture formation and low temperature reaction behavior were investigated using 2-D broadband chemiluminescence imaging, while high temperature ignition and combustion chemistry were studied using OH chemiluminescence imaging. The optical measurements were combined with cylinder pressure-based combustion analysis, including ignition delay and premixed burn duration, to validate the global behavior of the surrogate. Engine-out UHC, NO and soot emissions were also compared at different intake conditions, injection pressures and injection strategies. The proposed approach can provide validation data for further numerical engine combustion modeling and kinetic mechanism validation. The results show the surrogate was able to match the spray behavior, high temperature radical distribution, high temperature ignition delay, and the premixed burn duration of the target Jet-A fuel. Reasonable agreement was observed between low temperature ignition delay and radical distribution. The surrogate also captures the trend of the effects of intake conditions and injection pressure when single or double injections are used.
2014
- Photoionization Mass Spectrometric Measurements of Initial Reaction Pathways in Low-Temperature Oxidation of 2,5-DimethylhexaneBrandon Rotavera, Judit Zádor, Oliver Welz, Leonid Sheps, Adam M. Scheer, and 7 more authorsThe Journal of Physical Chemistry A, Nov 2014
Product formation from R + O2 reactions relevant to low-temperature autoignition chemistry was studied for 2,5-dimethylhexane, a symmetrically branched octane isomer, at 550 and 650 K using Cl-atom initiated oxidation and multiplexed photoionization mass spectrometry (MPIMS). Interpretation of time- and photon-energy-resolved mass spectra led to three specific results important to characterizing the initial oxidation steps: (1) quantified isomer-resolved branching ratios for HO2 + alkene channels; (2) 2,2,5,5-tetramethyltetrahydrofuran is formed in substantial yield from addition of O2 to tertiary 2,5-dimethylhex-2-yl followed by isomerization of the resulting ROO adduct to tertiary hydroperoxyalkyl (QOOH) and exhibits a positive dependence on temperature over the range covered leading to a higher flux relative to aggregate cyclic ether yield. The higher relative flux is explained by a 1,5-hydrogen atom shift reaction that converts the initial primary alkyl radical (2,5-dimethylhex-1-yl) to the tertiary alkyl radical 2,5-dimethylhex-2-yl, providing an additional source of tertiary alkyl radicals. Quantum-chemical and master-equation calculations of the unimolecular decomposition of the primary alkyl radical reveal that isomerization to the tertiary alkyl radical is the most favorable pathway, and is favored over O2-addition at 650 K under the conditions herein. The isomerization pathway to tertiary alkyl radicals therefore contributes an additional mechanism to 2,2,5,5-tetramethyltetrahydrofuran formation; (3) carbonyl species (acetone, propanal, and methylpropanal) consistent with β-scission of QOOH radicals were formed in significant yield, indicating unimolecular QOOH decomposition into carbonyl + alkene + OH.
- Fast exploration of chemical reaction networksPaolo Elvati and Angela VioliNov 2014
A variety of natural phenomena comprises a huge number of competing reactions and short-lived intermediates. Any study of such processes requires the discovery and accurate modeling of their underlying reaction network. However, this task is challenging due to the complexity in exploring all the possible pathways and the high computational cost in accurately modeling a large number of reactions. Fortunately, very often these processes are dominated by only a limited subset of the network’s reaction pathways. In this work we propose a novel computationally inexpensive method to identify and select the key pathways of complex reaction networks, so that high-level ab-initio calculations can be more efficiently targeted at these critical reactions. The method estimates the relative importance of the reaction pathways for given reactants by analyzing the accelerated evolution of hundreds of replicas of the system and detecting products formation. This acceleration-detection method is able to tremendously speed up the reactivity of uni- and bimolecular reactions, without requiring any previous knowledge of products or transition states. Importantly, the method is efficiently iterative, as it can be straightforwardly applied for the most frequently observed products, therefore providing an efficient algorithm to identify the key reactions of extended chemical networks. We verified the validity of our approach on three different systems, including the reactivity of t-decalin with a methyl radical, and in all cases the expected behavior was recovered within statistical error.
- A surrogate for emulating the physical and chemical properties of conventional jet fuelDoohyun Kim, Jason Martz, and Angela VioliCombustion and Flame, Nov 2014
Two surrogates are developed using a model-based optimizer to emulate the fuel properties affecting the spray development and gas phase ignition of a conventional jet fuel. The first surrogate, UM1, is a mixture of n-dodecane/iso-cetane/methylcyclohexane/toluene (0.3844/0.1484/0.2336/0.2336mol fraction), while the second, UM2, is a mixture of n-dodecane/iso-cetane/decalin/toluene (0.2897/0.1424/0.3188/0.2491mol fraction). The developed surrogates contain hydrocarbon species that are available in existing chemical mechanisms, and emulate both the physical and chemical properties of a representative real jet fuel, Jet-A POSF-4658. POSF-4658 is suitable as a surrogate target fuel since the properties of POSF-4658 are representatives of a nominal Jet-A, and a wide range of experimental POSF-4658 data is readily available, including multiple ignition delay measurements. While the UM1 surrogate gives a much tighter match for temperature-independent properties, UM2 is shown to better emulate liquid density and volatility, properties which are known to be important to spray predictions under engine relevant conditions. The new UM surrogates improve upon emulation of the chemical and physical properties of POSF-4658 compared with surrogates that currently exist in the literature. Ignition delay times predicted with a detailed chemical mechanism and the newly developed surrogate compositions under diesel relevant conditions show reasonable agreement with shock tube and rapid compression machine experiments.
- Stochastic atomistic simulation of polycyclic aromatic hydrocarbon growth in combustionJason Y. W. Lai, Paolo Elvati, and Angela VioliPhys. Chem. Chem. Phys., Nov 2014
Nanoparticles formed in gas phase environments, such as combustion, have an important impact on society both as engineering components and hazardous pollutants. A new software package, the Stochastic Nanoparticle Simulator (SNAPS) was developed, applying a stochastic chemical kinetics methodology, to computationally investigate the growth of nanoparticle precursors through trajectories of chemical reactions. SNAPS was applied to characterize the growth of polycyclic aromatic hydrocarbons (PAHs), important precursors of carbonaceous nanoparticles and soot, in a premixed laminar benzene flame, using a concurrently developed PAH growth chemical reaction mechanism, as well as an existing benzene oxidation mechanism. Simulations of PAH ensembles successfully predicted existing experimentally measured data and provided novel insight into chemical composition and reaction pathways. The most commonly observed PAH isomers in simulations showed the importance of 5-membered rings, which contrasts with traditionally assumed compositions involving primarily pericondensed 6-membered rings. In addition, the chemical growth of PAHs involved complex sequences of highly reversible reactions, rather than relatively direct routes of additions and ring closures. Furthermore, the most common reactions involved 5-membered rings, suggesting their importance to PAH growth. The framework developed in this work will facilitate future investigation of particle inception and soot formation and will benefit engineering of novel combustion technologies to mitigate harmful emissions.
2013
- Reaction Pathways for the Thermal Decomposition of Methyl ButanoateMohamad Akbar Ali and Angela VioliThe Journal of Organic Chemistry, Jan 2013
In recent years, biodiesel fuels, consisting of long-chain alkyl (methyl, ethyl, propyl) esters, have emerged as viable alternatives to petroleum-based fuels. From a combustion chemistry standpoint, there is great interest in developing accurate reaction models for these new molecules that can be used to predict their behaviors in various regimes. In this paper, we report a detailed study of the unimolecular decomposition pathways of methyl butanoate (MB), a short-chain ester that contains the basic chemical structure of biodiesel fuels. Using ab initio/DFT methods, we identified five homolytic fissions of C–C and C–O bonds and five hydrogen transfer reactions. Rate constants were determined using the G3B3 theory coupled with both variational transition state theory and Rice–Ramsperger–Kassel–Marcus/master equation simulations with hindered rotation corrections. Branching ratios in the temperature range 1500–2200 K indicate that the main pathway for thermal decomposition of MB is the reaction CH3CH2CH2C(═O)OCH3 \textrightarrow C2H5 + CH2C(═O)OCH3. The results, in terms of reaction pathways and rate constants, can be used for future development of mechanisms for long alkyl-chain esters.
- Thermodynamics of poly-aromatic hydrocarbon clustering and the effects of substituted aliphatic chainsPaolo Elvati and Angela VioliProceedings of the Combustion Institute, Jan 2013
Over the last few decades, the understanding of the processes related to the formation of soot has progressed considerably. However, the mechanisms that are responsible for the nucleation of soot are still unclear. While there is consensus that the formation of soot nuclei can be related to two classes of mechanisms (physical and chemical growth), their relative importance is still under debate. In particular, the aggregation of polycyclic aromatic hydrocarbons (PAHs), especially pyrene, has been proposed as key step for soot formation but strong experimental or computational proofs are still missing. To shed light on this issue, we conducted a thermodynamic analysis of the physical growth of poly-aromatic hydrocarbons using atomistic models. Free energy profiles of dimerization and trimerization processes of several PAHs are computed using molecular dynamics simulations in conjunction with advanced sampling techniques. Our study focuses not only on the potential energy of the clustering processes, but it also addresses the entropic contributions that affect the dimerization and trimerization of PAHs. The results of these simulations show that even at 1000K, only the formation of dimers of relative big species, such as ovalene or bigger, are favored over their corresponding free monomers, ruling out the simple stacking of pyrene as the main step for soot formation. The shapes of the free energy profiles also illustrate that there are no barriers in the exploration of the phase space and therefore, that the dimerization process itself is not kinetically controlled. Geometrical factors, such as the symmetry of the monomers, as well as the presence of substituted aliphatic chains play an important role in the physical agglomeration of PAH that can eventually lead to soot nuclei.
- PAH dimerization as the first step to soot particle inceptionJ Lowe, Paolo Elvati, and Angela VioliIn , Jan 2013
Particle inception, or the transition from the gas-phase to solid-phase, is a complex process that influences the growth of combustion-emitted particles, especially their size distribution and morphology. Unfortunately, although it is widely believed that nanoparticles form via polycyclic aromatic hydrocarbons (PAHs), little is known about the exact molecular mechanisms involved in the process. In this paper, we investigate the effects of molecular structure on the tendency of PAHs to dimerize. We measured the thermodynamic stability of aliphatic-substituted PAHs by calculating the free energy surfaces of monomer-monomer interactions. Specifically, we varied the geometry and chain lengths of these PAHs. Free energy was chosen as the best measure of thermodynamic stability because it takes into account stabilization due to both potential and entropic effects. The results show that the stability of aliphatic-substituted PAH dimers increases with the length and saturation of the aliphatic chains, resulting in lower free energy minima than their unsaturated analogs. The results can be used to expand the current soot models to include various species for the particle inception process.
- Studies of laminar opposed-flow diffusion flames of acetylene at low-pressures with photoionization mass spectrometryS.A. Skeen, B. Yang, H.A. Michelsen, J.A. Miller, A. Violi, and 1 more authorProceedings of the Combustion Institute, Jan 2013
We have designed an opposed-flow flame system to investigate the chemical composition of non-premixed flames using in situ flame-sampling molecular-beam mass spectrometry with synchrotron-generated tunable vacuum-ultraviolet light as an ionization source. This paper provides details of the experimental apparatus, sampling method, and data-reduction procedures. To test the system, we have investigated the chemical composition of three low-pressure (30–50Torr), non-premixed, opposed-flow acetylene(Ar)/O2(Ar) flames. We measured quantitative mole-fraction profiles as a function of the distance from the fuel outlet for the major species and several intermediates, including the methyl and propargyl radicals. We determined the temperature profiles of these flames by normalizing a sampling-instrument function to thermocouple measurements near the fuel outlet. A comparison of the experimental temperature and major species profiles with modeling results indicates that flame perturbations caused by the sampling probe are minimal. The observed agreement between experimental and modeled results, apparent for most combustion species, is similar to corresponding studies of premixed flames.
- Near-threshold photoionization mass spectra of combustion-generated high-molecular-weight soot precursorsScott A. Skeen, Hope A. Michelsen, Kevin R. Wilson, Denisia M. Popolan, Angela Violi, and 1 more authorJournal of Aerosol Science, Jan 2013
In this work, we present mass spectra showing organic species with mass-to-charge ratios between 15 and 900 sampled from near-atmospheric pressure, non-premixed, opposed-flow flames of acetylene, ethylene, and propane using an aerosol mass spectrometer with flash vaporization. Near-threshold photoionization was achieved by synchrotron-generated tunable vacuum-ultraviolet (VUV) light. Among the three different fuels, we observed variation in the mass progression, peak intensities, and isomeric content identifiable in photoionization-efficiency curves. The results indicate that different pathways contribute to the molecular growth of soot precursors and that the significance of these mechanisms is likely to depend on the fuel structure and/or flame conditions. Previous work has highlighted thermodynamic propensities for precursor formation; however, our results suggest that kinetic mechanisms play a role in determining the partitioning of soot precursor isomers under the conditions investigated here. Evidence for aliphatic-bridged and oxygenated species was also observed. Such species have been proposed as a possible precursor to particle inception following cluster formation but have never been confirmed.
2012
- Free Energy Calculation of Permeant-Membrane Interactions Using Molecular Dynamics SimulationsPaolo Elvati and Angela VioliJan 2012
Nanotoxicology, the science concerned with the safe use of nanotechnology and nanostructure design for biological applications, is a field of research that has recently received great attention, as a result of the rapid growth in nanotechnology. Many nanostructures are of a scale and chemical composition similar to many biomolecular environments, and recent papers have reported evident toxicity of selected nanoparticles. Molecular simulations can help develop a mechanistic understanding of how structural properties affect bioactivity. In this chapter, we describe how to compute the free energy of interactions between cellular membranes and benzene, the main constituent of some toxic carbonaceous particles, with well-tempered metadynamics. This algorithm reconstructs the free energy surface and accelerates rare events in a coarse-grained representation of the system.
- The role of the methyl ester moiety in biodiesel combustion: A kinetic modeling comparison of methyl butanoate and n-butaneKuang C. Lin, Jason Y.W. Lai, and Angela VioliFuel, Jan 2012
Growth of the biodiesel industry has motivated increased study of the combustion characteristics of its constituent molecules and building combustion modeling capability. Understanding how these characteristics differ between bio-derived and conventional diesel fuels can help in evaluating biodiesel performance. A kinetic modeling comparison of methyl butanoate and n-butane, its corresponding alkane, contrasted the combustion of methyl esters and normal alkanes, towards understanding the effect of the methyl ester moiety. Utilizing a combined n-heptane and methyl butanoate kinetic mechanism in shock tube simulations, the results predicted no region of negative temperature coefficient (NTC) behavior for methyl butanoate, compared to a well defined NTC region for n-butane. We observed that oxidation pathways associated with the methyl ester moiety inhibited NTC behavior, through increased production of hydroperoxy radicals (HO2) instead of hydroxyl radicals (OH). In addition, we compared the evolution of carbon monoxide, carbon dioxide, ethylene and acetylene. The early formation of CO and CO2, directly from methyl butanoate, revealed unique reaction pathways that also influenced a reduction in soot precursor formation. Overall, these results will help to understand how combustion processes change with the inclusion of oxygenated fuels, which will inform the study and design of combustion technologies.
2011
- Effect of Molecular Configuration on Binary Diffusion Coefficients of Linear AlkanesKyungchan Chae, Paolo Elvati, and Angela VioliThe Journal of Physical Chemistry B, Jan 2011
Mass diffusion coefficients are critically related to the predictive capability of computational combustion modeling. To date, the most common approach used to determine the molecular transport of gases is the Boltzmann transport equation of the gas kinetic theory. The Chapman-Enskog (CE) solution of this transport equation, combined with Lennard-Jones potential parameters, suggests a simple analytical expression for computing self and mutual diffusion coefficients. This approach has been applied over a wide range of flame modeling conditions due to its minimal computational requirement, despite the fact that the theory was developed only for molecules that have a spherical structure. In this study, we computed the binary diffusion coefficients of linear alkanes using all-atom molecular dynamics simulations over the temperature range 500-1000 K. The effect of molecular configurations on diffusion coefficients was determined relating the radii of gyration of the molecules to their corresponding collision diameters. The comparison between diffusion coefficients determined with molecular dynamics and the values obtained from the CE theory shows significant discrepancies, especially for nonspherical molecules. This study reveals the inability of CE theory with spherical potentials to account for the effect of molecular shapes on diffusion coefficients.
- Mutual diffusion coefficients of heptane isomers in nitrogen: A molecular dynamics studyKyungchan Chae and Angela VioliThe Journal of Chemical Physics, Jan 2011
The accurate knowledge of transport properties of pure and mixture fluids is essential for the design of various chemical and mechanical systems that include fluxes of mass, momentum, and energy. In this study we determine the mutual diffusion coefficients of mixtures composed of heptane isomers and nitrogen using molecular dynamics (MD) simulations with fully atomistic intermolecular potential parameters, in conjunction with the Green–Kubo formula. The computed results were compared with the values obtained using the Chapman–Enskog (C–E) equation with Lennard-Jones (LJ) potential parameters derived from the correlations of state values: MD simulations predict a maximum difference of 6\\% among isomers while the C–E equation presents that of 3\\% in the mutual diffusion coefficients in the temperature range 500–1000 K. The comparison of two approaches implies that the corresponding state principle can be applied to the models, which are only weakly affected by the anisotropy of the interaction potentials and the large uncertainty will be included in its application for complex polyatomic molecules. The MD simulations successfully address the pure effects of molecular structure among isomers on mutual diffusion coefficients by revealing that the differences of the total mutual diffusion coefficients for the six mixtures are caused mainly by heptane isomers. The cross interaction potential parameters, collision diameter \\\σ_\{12\} \\\ensuremathσ12, and potential energy well depth \\\\\varepsilon _\{12\}\\\\varepsilon12 of heptane isomers and nitrogen mixtures were also computed from the mutual diffusion coefficients.
- Peri-condensed aromatics with aliphatic chains as key intermediates for the nucleation of aromatic hydrocarbonsSeung-Hyun Chung and Angela VioliProceedings of the Combustion Institute, Jan 2011
Soot nucleation bridges the transition from gaseous hydrocarbons to macromolecular building blocks (nanoparticles) that eventually turn into soot. Polycyclic aromatic hydrocarbons (PAH) have often been invoked as important compounds of this process but their role has not been clearly identified. In this paper we report on a detailed analysis of the physical interactions between PAH in the range 200–450amu using Molecular Dynamics simulations. In particular, we identified a pool of nine aromatics and studied their clustering behaviors in systems composed of thousands of homo-molecular and hetero-molecular molecules to understand the influence of molecular mass, morphology and temperature on the nucleation process. At temperatures higher than 1000K, small clusters of PAH (2–5 molecules) are detected but they are not stable enough to accommodate the further growth into larger particles. This result raises doubts on the ability of these molecules to become soot nuclei. Molecular morphology is another important parameter for the nucleation process. Aromatics with attached aliphatic chains show considerably faster nucleation rates than the corresponding polycyclic aromatic hydrocarbons of similar mass without any chain. The collision efficiency is not increased by the aliphatic chain attachments, which may indicate that a faster nucleation process for these systems is due to the ability of these molecules to accommodate the collision energy into additional internal vibrational modes of the aliphatic chains. The results of this study provide information on the clustering behavior of PAH and can lead to the development of a more complex model to describe the physical nucleation of PAH that includes molecular masses, morphologies and temperature as main parameters to describe the transition from gas-phase species to macromolecular structures.
- Biodiesel combustion: Advances in chemical kinetic modelingJason Y.W. Lai, Kuang C. Lin, and Angela VioliProgress in Energy and Combustion Science, Jan 2011
Burgeoning global demand for energy has increased concerns about the fuel security issues and deleterious environmental impacts that result from the ubiquitous use of fossil fuels to meet these needs. This article is a review of completed work towards the goal of creating chemical kinetic mechanisms for biodiesel, which will aid in the development of clean and efficient combustors that utilize alternative fuels. As the composition of biodiesel is too complex to directly model, efforts have instead focused on the development of mechanisms for surrogates, simpler molecules that can produce the primary characteristics of biodiesel combustion. Research initially targeted smaller molecules like methyl butanoate to investigate the role of the characteristic ester group that is present in the fatty acid alkyl esters that comprise biodiesel. The study of isomers and similar unsaturated compounds elucidated the effects of molecular structure on combustion. Subsequent efforts involved the study of larger molecules that are close in scale to biodiesel molecules, such as methyl decanoate, as well as molecules that are present in biodiesel, such as methyl stearate. Applications of kinetic modeling demonstrate its utility in the study of combustion through, for example, revealing the chemistry in the early formation of CO2 in biodiesel and its soot reduction tendencies. The results of this review illustrate key limitations in kinetic modeling, namely a need for high-pressure kinetic methodology and a need for continuous improvement of kinetic mechanisms through theory and experiment. These limitations suggest direction for future research; further experimental and theoretical work will produce accurate mechanisms for appropriate biodiesel surrogates. All of these efforts represent significant advances in kinetic modeling that are important towards the goal of building a predictive capability for biodiesel combustion. Such predictive capability will aid the development of combustion technologies that will help society meet its energy needs in an environmentally conscious manner.
2010
- Nucleation of fullerenes as a model for examining the formation of sootSeung Hyun Chung and Angela VioliThe Journal of Chemical Physics, May 2010
The formation of soot begins with the nucleation of nanoparticles, a process difficult to model due to the complexity of the constituent particles. Fullerenes have characteristics resembling the particles found in soot, but their simpler structure makes simulations more tractable. We propose that the nucleation of fullerenes may serve as a window to the formation of soot nuclei. Using molecular dynamics simulations, we analyze the nucleation rates of homomolecular systems of C60, C80, C180, and C240 fullerenes as function of temperature and molecular mass. For temperatures lower than 1000 K, the four systems show similar characteristics, with significant nucleation rates, due to the low energy that favors binding. At higher temperatures, the high kinetic energy limits the binding probability between fullerenes, and molecular clusters are only detected in systems composed of C180 and C240. The analysis shows that particles with molecular masses between those of C80 and C180 could be critical for the transition from monomers to clusters. The computational findings are then related to experimental data of combustion-generated particles present in the literature to assess the feasibility of a physical nucleation pathway in high temperature regimes. The results obtained using molecular dynamics simulations highlight the importance of a physical nucleation pathway to describe the formation of molecular clusters when the particle concentration exceeds a critical value. These results represent the first step toward a more complete description of nanoparticle formation and soot nucleation in high temperature regimes.
- Pyrolytic Hydrocarbon Growth from CyclopentadieneDo Hyong Kim, James A. Mulholland, Dong Wang, and Angela VioliThe Journal of Physical Chemistry A, Dec 2010
Aromatic hydrocarbon growth from cyclopentadiene (CPD) was studied using a laminar flow reactor operating in the temperature range 550-950 \textdegreeC without oxygen. Benzene, indene, and naphthalene were the major products, which is in agreement with the previous computational studies on the reaction pathways from CPD. A crossover of indene and naphthalene yields around 775 \textdegreeC was also observed, which further supports the results of the computational studies. Although the specific intermediates in the proposed pathways from CPD were not detected, the high selectivity of products and the observation of other methylindene and dihydronaphthalene intermediates suggest that the recombination of two CPDs via radical-molecule and/or radical-radical pathways to form indene and naphthalene is the dominant formation pathway. In addition to the products from the CPD-CPD reactions, the products from the reactions of CPD with indene, naphthalene, and acenaphthylene were also observed, which demonstrate the importance of CPD in carbon growth.
- Simulation of Nanoparticle Permeation through a Lipid MembraneSteven L. Fiedler and Angela VioliBiophysical Journal, Dec 2010
A metric of nanoparticle toxicity is the passive permeability rate through cellular membranes. To assess the influence of nanoparticle morphology on this process, the permeability of buckyball-sized molecules through a representative lipid bilayer was investigated by molecular-dynamics simulation. When C60 was compared with a prototypical opened C60 molecule and a representative combustion-generated particle, C68H29, the calculated free-energy profiles along the permeation coordinate revealed a sizable variation in form and depth. The orientation of the anisotropic molecules was determined by monitoring the principal axis corresponding to the largest moment of inertia, and free rotation was shown to be hindered in the bilayer interior. Diffusion constant values of the permeant molecules were calculated from a statistical average of seven to 10 trajectories at five locations along the permeation coordinate. A relatively minor variation of the values was observed in the bilayer interior; however, local resistance values spanned up to 24 orders of magnitude from the water layer to the bilayer center, due primarily to its exponential dependence on free energy. The permeability coefficient values calculated for the three similarly sized but structurally distinct nanoparticles showed a significant variance. The use of C60 to represent similarly sized carbonaceous nanoparticles for assessments of toxicity is questioned.
- Natural convection heat transfer of nanofluids in a vertical cavity: Effects of non-uniform particle diameter and temperature on thermal conductivityKC Lin and Angela VioliDec 2010
This paper analyzes the heat transfer and fluid flow of natural convection in a cavity filled with Al2O3/water nanofluid that operates under differentially heated walls. The Navier-Stokes and energy equations are solved numerically, coupling Xu’s model (Xu et al., 2006) for calculating the effective thermal conductivity and Jang’s model (Jang et al., 2007) for determining the effective dynamic viscosity, with the slip mechanism in nanofluids. The heat transfer rates are examined for parameters of non-uniform nanoparticle size, mean nanoparticle diameter, nanoparticle volume fraction, Prandtl number, and Grashof number. Enhanced and mitigated heat transfer effects due to the presence of nanoparticles are identified and highlighted. Based on these insights, we determine the impact of fluid temperature on the heat transfer of nanofluids. Decreasing the Prandtl number results in amplifying the effects of nanoparticles due to increased effective thermal diffusivity. The results highlight the range where the heat transfer uncertainties can be affected by the size of the nanoparticles. \textcopyright 2009 Elsevier Inc. All rights reserved.
2009
- Order-Disorder Phase Transformation of Triacylglycerols: Effect of the Structure of the Aliphatic ChainsWen-Dung Hsu and Angela VioliThe Journal of Physical Chemistry B, Jan 2009
Plant oils have been used as environmentally benign lubricants since they present high viscosity index and flash points and low evaporation loss. Triacylglycerols (TAG) are the major components of naturally occurring oils and fats and are able to produce high strength lubricant films. One of the main concerns that hinders the usage of triacylglycerols as lubricants, however, is the thermal stability of these molecules. In this paper, we report on the effect of chain structure on density, viscosity, and thermal stability of triacylglycerols using molecular dynamics simulations. The selected triacylglycerols are trilauroylglycerol (LLL-TAG), tristearoylglycerol (SSS-TAG), trans-trioleoylglycerol (trans-OOO-TAG), and trans-trilinolenoylglycerol (trans-LeLeLe-TAG). The first two TAGs are saturated molecules with a different number of carbons in the chain, and the second two TAGs are monounsaturated and polyunsaturated molecules, respectively. The computed results demonstrate that the length of the aliphatic chain influences the physical properties of triacylglycerols. TAGs with short chain (LLL-TAG) show higher density than TAGs with longer chains. Viscosity is determined by the degree of recoil of the aliphatic chains and by the number and location of unsaturated bonds. Thermal stability, as represented by the ability of triacylglycerols to stay in a disordered phase during the cooling process, is related to the order-disorder phase transition temperature. Since the phase transition temperature can be correlated to the thermal stability during the cooling process, LeLeLe-TAG shows the highest thermal stability among the systems considered. These results can aid in the design of molecules with specific lubrication properties.
- An experimental and computational study of methyl ester decomposition pathways using shock tubesA. Farooq, D.F. Davidson, R.K. Hanson, L.K. Huynh, and A. VioliProceedings of the Combustion Institute, Jan 2009
The high-temperature decomposition of three simple methyl esters: methyl acetate, methyl propionate and methyl butanoate, were studied behind reflected shock waves using tunable diode laser absorption of CO2 near 2.7\ensuremathμm. CO2 yield measurements were made over the range of temperatures 1260–1653K, pressures of 1.4–1.7atm and reactant concentrations of 2–3%, with the balance Ar. The CO2 absorption strengths near 2.7\ensuremathμm are approximately 50 to 1000 times stronger than the bands near 2.0 and 1.55\ensuremathμm, respectively, and offer opportunities for significantly more sensitive and accurate combustion measurements than previous absorption work using CO2 bands at shorter wavelength. The experiments provide the first laser-based time-history measurements of the CO2 yields during pyrolysis of these bio-diesel surrogate fuels in a shock tube. Model predictions for CO2 yields during methyl butanoate pyrolysis at high temperatures, using the detailed reaction mechanisms of [E. M. Fisher, W. J. Pitz, H. J. Curran, C. K. Westbrook, Proc. Combust. Inst. 28 (2000) 1579-1586.] and others, are significantly lower than those measured in this study. However, an improved methyl butanoate model which extends the recent theoretical work of [L.K. Huynh, A. Violi, J. Org. Chem. 73 (2008) 94–101.] provides substantially improved predictions of CO2 yields during methyl butanoate pyrolysis. As earlier mechanisms predicted low yields of CO2 from methyl butanoate decomposition, these new findings imply that existing bio-diesel fuel models, which rely on the rapid formation of two oxygenate radicals from methyl esters (rather than a single non-reactive CO2 molecule) to account for the tendency for soot reduction, may have to be revisited.
- Simulating carbonaceous pollutant nanoparticles an aid to discoveryS Fiedler and Angela VioliJan 2009
An international consortium and EPA specified a set of prototypical nanomaterials such as carboneous molecules for toxicological testing to address concerns regarding public reaction to environmental risk assessment programs and a subsequent call for standardization of nanoparticle toxicity measurement techniques. A multiscale approach developed by the Violi group is designed to provide the necessary computational tools to tackle simulation of mesoscopic-sized systems. FORTRAN-based Atomistic Model Particle Inception (AMPI) code is able to create designer fuel additives that could shift the PM size distribution outside the range found to be most toxic. Simulation studies found that the presence of a permeated nanoparticle is found to hinder the motion of the surrounding lipid and cholesterol molecules, acting as an anti-plasticizer in the lipid bilayers.
2008
- Science-based model for particle formation from novel fuelsA VioliJournal of Physics: Conference Series, Jul 2008
With the advent of petascale high-performance computing platforms, realistic multiscale modeling can be constructed to incorporate atomic-scale (molecular) information into macroscopic predictions of engineering systems. The overriding theme of the work presented in this paper is developing a multiscale modeling approach for soot formulation where atomistic data is integrated into macroscopic simulations. The prediction of soot formation remains arguably one of the most challenging subjects in combustion science, having an influence over a wide range of applications ranging from combustion efficiency to reducing emissions to slow global warming, to improved heat transfer designs in industrial settings, to predicting the radiation heat transfer from large scale fires. Starting from the fuel structures the new multiscale simulations reveals how chemical changes and transformation can propagate upward in scale to help define the function of the particle structures. In particular, the fuel structure influences the morphology of the nanoparticles, which in turn is critical in determining the overall growth and agglomeration behavior. These simulations make use of a newly proposed combination of molecular dynamics and kinetic Monte Carlo methodologies that will include both chemical reactions and agglomeration processes. The main strength of this approach is the ability to use important atomic-scale information directly into large scale description of the macroscopic phenomena.
- Kinetics Study of the OH + Alkene \textrightarrow H2O + Alkenyl Reaction ClassLam K. Huynh, Kyle Barriger, and Angela VioliThe Journal of Physical Chemistry A, Feb 2008
In this paper we report on the kinetics of hydrogen abstraction for the OH + alkene reaction class, using the reaction class transition state theory (RC-TST) combined with the linear energy relationship (LER) and the barrier height grouping (BHG) approaches. Parameters for the RC-TST were derived from theoretical calculations using a set of 15 reactions representing the hydrogen abstractions from the terminal and nonterminal carbon sites of the double bond of alkene compounds. Both the RC-TST/LER, where only reaction energy is needed at either density functional theory BH&HLYP or semiempirical AM1 levels, and RC-TST/BHG, where no additional information is required, are found to be promising methods for predicting rate constants for a large number of reactions in this reaction class. Detailed error analyses show that, when compared to explicit theoretical calculations, the averaged systematic errors in the calculated rate constants using both the RC-TST/LER and RC-TST/BHG methods are less than 25% in the temperature range 300-3000 K. The estimated rate constants using these approaches are in good agreement with available data in the literature.
- Kinetic Modeling of Methyl Butanoate in Shock TubeLam K. Huynh, Kuang C. Lin, and Angela VioliThe Journal of Physical Chemistry A, Dec 2008
An increased necessity for energy independence and heightened concern about the effects of rising carbon dioxide levels have intensified the search for renewable fuels that could reduce our current consumption of petrol and diesel. One such fuel is biodiesel, which consists of the methyl esters of fatty acids. Methyl butanoate (MB) contains the essential chemical structure of the long-chain fatty acids and a shorter, but similar, alkyl chain. This paper reports on a detailed kinetic mechanism for MB that is assembled using theoretical approaches. Thirteen pathways that include fuel decomposition, isomerization, and propagation steps were computed using ab initio calculations [J. Org. Chem. 2008, 73, 94]. Rate constants from first principles for important reactions in CO2 formation, namely CH3OCO═CH3 + CO2 (R1) and CH3OCO═CH3O + CO (R2) reactions, are computed at high levels of theory and implemented in the mechanism. Using the G3B3 potential energy surface together with the B3LYP/6-31G(d) gradient, Hessian and geometries, the rate constants for reactions R1 and R2 are calculated using the Rice-Ramsperger-Kassel-Marcus theory with corrections from treatments for tunneling, hindered rotation, and variational effects. The calculated rate constants of reaction R1 differ from the data present in the literature by at most 20%, while those of reaction R2 are about a factor of 4 lower than the available values. The new kinetic model derived from ab initio simulations is combined with the kinetic mechanism presented by Fisher et al. [Proc. Combust. Inst. 2000, 28, 1579] together with the addition of the newly found six-centered unimolecular elimination reaction that yields ethylene and methyl acetate, MB = C2H4 + CH3COOCH3. This latter pathway requires the inclusion of the CH3COOCH3 decomposition model suggested by Westbrook et al. [Proc. Combust. Inst. 2008, accepted]. The newly composed kinetic mechanism for MB is used to study the CO2 formation during the pyrolysis of MB as well as to investigate the autoignition of MB in a shock tube reactor at different temperatures and pressures. The computed results agree very well with experimental data present in the literature. Sensitivity and flux (rate-of-production) analyses are carried out for the CO2 formation with the new MB mechanism, together with available reaction mechanisms, to assess the importance of various kinetic pathways for each regime. With the new mechanism, the flux analyses for the formation of C2H species, one of the most important species for ignition delay time, are also presented at different conditions. In addition to giving a better chemical insight of the pyrolysis/oxidation of MB, the results suggest ways to improve the mechanism’s capability to predict CO2 formation and ignition delay times in pyrolysis and oxidation conditions.
- Molecular dynamics simulation study of a pulmonary surfactant film interacting with a carbonaceous nanoparticleSeungho Choe, Rakwoo Chang, Jonggu Jeon, and Angela VioliBiophys J, Nov 2008
This article reports an all-atom molecular dynamics simulation to study a model pulmonary surfactant film interacting with a carbonaceous nanoparticle. The pulmonary surfactant is modeled as a dipalmitoylphosphatidylcholine monolayer with a peptide consisting of the first 25 residues from surfactant protein B. The nanoparticle model with a chemical formula C188H53 was generated using a computational code for combustion conditions. The nanoparticle has a carbon cage structure reminiscent of the buckyballs with open ends. A series of molecular-scale structural and dynamical properties of the surfactant film in the absence and presence of nanoparticle are analyzed, including radial distribution functions, mean-square displacements of lipids and nanoparticle, chain tilt angle, and the surfactant protein B peptide helix tilt angle. The results show that the nanoparticle affects the structure and packing of the lipids and peptide in the film, and it appears that the nanoparticle and peptide repel each other. The ability of the nanoparticle to translocate the surfactant film is one of the most important predictions of this study. The potential of mean force for dragging the particle through the film provides such information. The reported potential of mean force suggests that the nanoparticle can easily penetrate the monolayer but further translocation to the water phase is energetically prohibitive. The implication is that nanoparticles can interact with the lung surfactant, as supported by recent experimental data by Bakshi et al.
- Thermal Decomposition of Methyl Butanoate:\thinspace Ab Initio Study of a Biodiesel Fuel SurrogateLam K. Huynh and Angela VioliThe Journal of Organic Chemistry, Jan 2008
In this paper, we report a detailed analysis of the breakdown kinetic mechanism for methyl butanoate (MB) using theoretical approaches. Electronic structures and structure-related molecular properties of reactants, intermediates, products, and transition states were explored at the BH&HLYP/cc-pVTZ level of theory. Rate constants for the unimolecular and bimolecular reactions in the temperature range of 300-2500 K were calculated using Rice-Ramsperger-Kassel-Marcus and transition state theories, respectively. Thirteen pathways were identified leading to the formation of small compounds such as CH3, C2H3, CO, CO2, and H2CO. For the initial formation of MB radicals, H, CH3, and OH were considered as reactive radicals participating in hydrogen abstraction reactions. Kinetic simulation results for a high temperature pyrolysis environment show that MB radicals are mainly produced through hydrogen abstraction reactions by H atoms. In addition, the C(O)OCH3 = CO + CH3O reaction is found to be the main source of CO formation. The newly computed kinetic sub-model for MB breakdown is recommended as a core component to study the combustion of oxygenated species.
- Coarse-Graining in Time: From Microscopics to MacroscopicsAngela VioliIn Coarse-Graining of Condensed Phase and Biomolecular Systems, Jan 2008
2007
- Thermal Decomposition of Decalin: An Ab Initio StudyKyungchan Chae and Angela VioliThe Journal of Organic Chemistry, Apr 2007
Density functional theory calculations (B3LYP and BH&HLYP functionals) of the potential energy surface have been performed to investigate the mechanisms of decalin breakdown, and the Rice-Ramsperger-Kassel-Marcus and transition state theory methods have been used to compute the high-pressure limit thermal rate constants for the new reaction pathways. The new pathways connect decalin to five primary monoaromatic species:\thinspace benzene, toluene, styrene, ethylbenzene, and xylene. The reactions used for the new routes are carbon-carbon bond cleavage reaction, dissociation reaction, and hydrogen abstraction and addition reactions. A kinetic analysis was performed for pyrolytic conditions, and benzene, toluene, and xylene were identified as major products.
- Insights on the nanoparticle formation process in counterflow diffusion flamesSeung Hyun Chung and Angela VioliCarbon, Apr 2007
This paper reports on a new version of the multiscale atomistic model for particle inception (AMPI) code to study the effect of oxidation on nanoparticle structures formed in counter-flow, propene diffusion flames. The code has been expanded with 158 new oxidation reactions and the potential for the Molecular Dynamics module has been expanded to describe systems containing C, H and O. The rate of nanoparticle mass growth is modeled in two different flame configurations to address the influence of combustion environments on the inception process. In the case of high temperature and abundant radicals, the AMPI code successfully predicts the nanoparticle formation. In these conditions, the particle loading is mainly associated with the formation of high-molecular structures through polymerization reactions. In the case of low temperature and high polycyclic aromatic hydrocarbons (PAH) concentrations, the combined mechanism of chemical reactions described by the AMPI code and coagulation of PAH and nanoparticles contribute to particle growth. Structural properties of the particles, such as aspect ratio, oxygen content and H/C ratio are computed in the two configurations.
- Nanoparticle formation in counter-diffusion flamesSH Chung and Angela VioliIn , Apr 2007
The microstructures of atmospheric pressure, counter-flow, propane diffusion flames were studied numerically using the multiscale Atomistic Model for Particle Inception (AMPI) code. For diffusion flames, flow characteristics played an important role in the formation and growth. The flame location could significantly affect the oxidation of particles and sooting characteristics. Counterflow diffusion flames were normally located on the oxidizer side, but upon appropriate dilution of the fuel and oxidizer streams, the flame could be moved to the fuel side. The formation of nanoparticles in different combustion regimes was also studied using the AMPI code to address the importance of oxidation on particle growth. This is an abstract of a paper presented at the 2007 AIChE Annual Meeting (Salt Lake City, UT 11/4-9/2007).
- The effect of temperature on nanoparticle clusteringSteven L. Fiedler, Sergei Izvekov, and Angela VioliCarbon, Apr 2007
A multiscale computational approach was applied to study the evolution of soot formation in an ethylene flame environment, from small polycyclic aromatic hydrocarbons of less than 10 atoms, to nano-sized organic carbon (NOC) clusters of diameters up to 15nm. As this evolution covers sizable breadth in time and length scales, the application of a sequence of complementary computational approaches was employed. To model NOC particle aggregation, the coarse grain method allowed for molecular dynamics simulation of over 13,000 nanoparticles (106 atoms), for hundreds of nanoseconds. Notable growth in both the number and size of particles was observed when temperature was dropped from 1800 to 300K. A test of input sensitivity determined little noticeable effect upon inclusion of electrostatic interactions of atomic-centered partial charges. These results provide insights on the influence of temperature on nanoparticle aggregation in flames. This could also assist in the analysis of experimental measurements of methods which allow for sample cooling in the period from extraction to detection.
- Ab initio study on the methyl butanoate decompositionLK Huynh and Angela VioliIn , Apr 2007
Methyl butanoate (MB) has been widely used as a convenient surrogate for biodiesel fuels in flame simulation. The potential energy surface for the identified main pathways of MB breakdown were studied using density functional theory methods, i.e., BH&HLYP and B3LYP. The RRKM and the Master Equation methods were used to derive pressure-dependent rate constants for unimolecular reactions in high-pressure-limit and atmospheric regime, respectively. The canonical transition state theory was also employed to calculate thermal rate constants for bimolecular reactions. The pattern of the MB decomposition was served as a prototype for identifying important breakdown pathways and assigning kinetic parameters for such pathways for low molecular weight triglycerides, e.g., tributyrin. This is an abstract of a paper presented at the 2007 AIChE Annual Meeting (Salt Lake City, UT 11/4-9/2007).
- Soot primary particle formation from multiscale coarse-grained molecular dynamics simulationAngela Violi and Sergei IzvekovProceedings of the Combustion Institute, Apr 2007
A new multiscale coarse-graining procedure is used to study carbonaceous nanoparticle agglomeration in combustion environments. The computational methodology is applied to an ensemble of 10,000 nanoparticles (or effectively 2 million total carbon atoms) to simulate, for the first time, the agglomeration of carbonaceous nanoparticles using coarse-grained atomistic-scale information. In particular, with the coarse-graining approach we are able to assess the influence of nanoparticle morphology and temperature on the agglomeration process. The coarse-graining of the interparticle force field is accomplished applying a force-matching procedure to data obtained from trajectories and forces from all-atom MD simulations. The coarse-grained MD results show rich and varied clustering behaviors for different particle morphology and, in some cases, the formation of primary particles with a diameter around 15nm are observed for the first time by molecular simulation techniques.
2006
- Combustion-generated nanoparticles produced in a benzene flame: A multiscale approachAngela Violi and Arun VenkatnathanThe Journal of Chemical Physics, Aug 2006
This paper details the multiscale methodology developed to analyze the formation of nanoparticles in a manner that makes it possible to follow the evolution of the structures in a chemically specific way. The atomistic model for particle inception code that combines the strengths of kinetic Monte Carlo and molecular dynamics is used to study the chemical and physical properties of nanoparticles generated in a premixed fuel-rich benzene flame, providing atomistic scale structures (bonds, bond angles, dihedral angles) as soot precursors evolve into a three-dimensional structure. Morphology, density, porosity, and other physical properties are computed. Two heights corresponding to two different times in the benzene flame, experimentally studied by Bittner and Howard [Proc. Combust. Inst.\hphantom,18, 1105 (1981)], were chosen to examine the influence of different environments on structural properties of the particles formed.
- A Coarse-Grained Molecular Dynamics Study of Carbon Nanoparticle AggregationSergei Izvekov and Angela VioliJournal of Chemical Theory and Computation, May 2006
A multiscale coarse-graining procedure is used to study carbonaceous nanoparticle assembly. The computational methodology is applied to an ensemble of 10\thinspace000 nanoparticles (or effectively 2 million total carbon atoms) to simulate the agglomeration of carbonaceous nanoparticles using coarse-grained atomistic-scale information. In particular, with the coarse-graining approach, we are able to assess the influence of nanoparticle morphology and temperature on the agglomeration process. The coarse-graining of the interparticle force field is accomplished applying a force-matching procedure to data obtained from trajectories and forces from all-atom molecular dynamics simulation. The coarse-grained molecular dynamics results show rich and varied clustering behaviors for different particle morphologies. They are shown to reproduce accurately the structural properties of the nanoparticles systems studied, while allowing for molecular dynamics simulations of much larger self-assembled nanoparticles systems.
- Formation of Naphthalene, Indene, and Benzene from Cyclopentadiene Pyrolysis:\thinspace A DFT StudyDong Wang, Angela Violi, Do Hyong Kim, and James A. MullhollandThe Journal of Physical Chemistry A, Apr 2006
Four new reaction pathways for polycyclic aromatic hydrocarbon growth from cyclopentadiene pyrolysis are proposed and investigated using the B3LYP/6-31G(d,p) level of theory. These pathways allow for the production of indene, naphthalene, and benzene through intramolecular addition, C-H β-scission, and C-C \β\-scission reaction mechanisms, respectively. Results show that the intramolecular addition channel is favored at low temperatures, and the C-H \β\-scission channel and the newly identified C-C \β\-scission pathway become significant when the temperature increases. These results are in qualitative agreement with the experimental results previously obtained by this research group indicating that the main product at low temperature is indene, while benzene and naphthalene production dominate at the high-temperature end.
- Insights into the Effect of Combustion-Generated Carbon Nanoparticles on Biological Membranes: A Computer Simulation StudyRakwoo Chang and Angela VioliThe Journal of Physical Chemistry B, Mar 2006
Classical molecular dynamics simulations of atomistic models of combustion-generated carbon nanoparticles and lipid bilayers have been performed to explore their possible structural, dynamical, and thermodynamic effects on biological membranes. The DREIDING generic force field is used for the carbonaceous nanoparticles of different morphologies, as produced from combustion sources, and the united atom model was employed for the dimyristoylphosphatidylcholine (DMPC) bilayer. It is observed that particle shape and structure have significant effects on solvation, mobility, adsorption, and permeation behavior of the particles. While combustion-generated carbon nanoparticles with an aspect ratio close to unity prefer to stay near the membrane center, precursors with other shapes mostly reside within the hydrocarbon tail region of the membrane. Carbon nanoparticles are not trapped in a local region even inside the membranes but move freely with a speed depending on their molecular weight. The adsorption of the particles on the surface of the biological membrane is comparable to thermal fluctuations because the weak segregation effect by water molecules is the main driving force to the adsorption behavior. The bigger the precursors are, the stronger they are bound to the membrane surface. The presence of combustion-generated nanoparticles inside the membrane perturbs local lipid density by pushing the neighboring lipid molecules away from the nanoparticles. This, coupled with thermal fluctuations, can induce an instantaneous membrane pore to allow water protrusion. From the umbrella sampling method, the potential of mean force for the permeation of carbona nanoparticles into the bilayer was also obtained. Surprisingly, elongated particles have a free energy barrier an order of magnitude smaller compared with more round ones. In addition, the round carbon nanoparticles showed strong hysteresis due to the local trapping of water molecules. Although the carbon soot precursors studied in this work are not the well-known carbon nanoparticles such as fullerenes or carbon nanotubes, the qualitative features of this study may be applicable to them as well.
- Radical-Molecule Reactions for Aromatic Growth: A Case Study for Cyclopentadienyl and AcenaphthyleneDong Wang and Angela VioliThe Journal of Organic Chemistry, Oct 2006
Polycyclic aromatic hydrocarbon growth from acenaphthylene and cyclopentadienyl was investigated by using the B3LYP/6-31G(d,p) and BH&HLYP/6-31G(d,p) levels of theory as well as transition state theory. The reaction pathways of cyclopentadienyl bearing hydrocarbons are different from those without these moieties and cannot be adequately accounted for by the existing acetylene addition and aryl-aryl addition mechanisms. The reaction mechanisms identified in this paper lead to the formation of fluoranthene, aceanthrylene, and acephenanthrylene. Rate constants of the radical-molecule addition and subsequent intramolecular addition steps predict that the 1,2 double bond in acenaphthylene is much more reactive than the 3,4 and 4,5 double bonds. Fluoranthene is the most abundant product produced at high temperatures and the yield of acephenanthrylene is bigger than that of aceanthrylene. The computational results are discussed in light of pyrolysis experiments on CPD-indene and CPD-acenaphthylene mixtures conducted by Prof. Mulholland’s research group reported in a previous work.
- An improvement in soot formation mechanism using modified HACA techniquesHR Zhang, Angela Violi, EG Eddings, and AF SarofimIn , Oct 2006
A new soot formation model using particle dynamics directly coupled with gas phase chemistry was evaluated on three ethylene and three methane laminar premixed flames. Using the modified moments techniques, the model included particle nucleation, coagulation, acetylene addition, surface condensation, and oxidation modes of particle dynamics. The model showed strength in the prediction of concentrations of major combustion products and critical intermediates, and characteristics of soot particles. The model extended the practicability of simulation beyond the prediction of soot volume fraction and estimated the mean particle diameter quite well for methane and ethylene flames with a wide range of C/O ratios. In particular, the model expanded the range of fuels that can be simulated to methane flames; it also correctly predicts C/O ratio effects on soot formation. This is an abstract of a paper presented at the 231st ACS National Meeting (Atlanta, GA 3/26-30/2006).
2005
- Systematic Coarse-Graining of Nanoparticle Interactions in Molecular Dynamics SimulationSergei Izvekov, Angela Violi, and Gregory A. VothThe Journal of Physical Chemistry B, Sep 2005
A recently developed multiscale coarse-graining procedure [Izvekov, S.; Voth, G. A. J. Phys. Chem. B 2005, 109, 2469] is extended to derive coarse-grained models for nanoparticles. The methodology is applied to C60 and to carbonaceous nanoparticles produced in combustion environments. The coarse-graining of the interparticle force field is accomplished applying a force-matching procedure to data obtained from trajectories and forces from all-atom MD simulations. The CG models are shown to reproduce accurately the structural properties of the nanoparticle systems studied, while allowing for MD simulations of much larger self-assembled nanoparticle systems.
- Cyclodehydrogenation Reactions to Cyclopentafused Polycyclic Aromatic HydrocarbonsAngela VioliThe Journal of Physical Chemistry A, Sep 2005
B3LYP/6-31G(d,p) electronic structure calculations are employed to elucidate the reaction mechanisms for the conversion of the alternant C18H12 polycyclic aromatic hydrocarbon benzo[c]phenanthrene into the nonalternant C18H10 PAHs cyclopenta[cd]pyrene and benzo[ghi]fluoranthene. Isomerization reactions such as 5/6-ring switching and hydrogen atom scrambling are analyzed. Bay region chemistry, involving the rupture of one benzene ring followed by the formation of a new five-membered ring, is also studied, together with the mechanism for the formation of an aryne. The rearrangement of the latter yields annelated cyclopentadienylidenecarbene, which is then trapped intramolecularly.
- Detailed Modeling of the Molecular Growth Process in Aromatic and Aliphatic Premixed FlamesA. D’anna and A. VioliEnergy & Fuels, Jan 2005
A detailed kinetic model has been developed and used to simulate aromatic growth in premixed benzene and ethylene flames. The model considers the role of resonantly stabilized radicals in the growth of aromatic species, in addition to the hydrogen abstraction carbon addition (HACA) mechanism, which involves hydrogen abstraction to activate aromatics followed by subsequent acetylene addition. Model results show that the self-combination of resonantly stabilized radicalsin particular, the combination of cyclopentadienyl radicalsis the controlling pathway for the aromatic-ring growth. The kinetic model reproduces the experimental trends of these compounds already below the flame front and their decrease within the flame front, which has been observed experimentally but never predicted numerically. The reaction mechanism has been used to identify the different behaviors of aromatic growth in ethylene and benzene flames. Benzene formation is the rate-limiting step for aromatic growth in ethylene flames. C6H6 is formed across the flame front and carbon growth continues through the formation of multi-ring aromatics in the post-flame region. In benzene oxidation, the aromatic ring is already present in the main oxidation zone and it is mainly oxidized to cyclopentadienyl radicals. As a consequence, a large amount of cyclopentadienyl radicals are available for recombination reactions, leading to multi-ring formation already in the main oxidation region. The increase of the temperature at the flame front reduces their concentration due to pyrolysis and/or oxidation resulting in a peak value in the main oxidation region and a leveling-off in the post-oxidation region of the flame.
- Computational and experimental study of JP-8, a surrogate, and its components in counterflow diffusion flamesJames A. Cooke, Matteo Bellucci, Mitchell D. Smooke, Alessandro Gomez, Angela Violi, and 2 more authorsProceedings of the Combustion Institute, Jan 2005
Non-sooting counterflow diffusion flames have been studied both computationally and experimentally, using either JP-8, or a six-component JP-8 surrogate mixture, or its individual components. The computational study employs a counterflow diffusion flame model, the solution of which is coupled with arc length continuation to examine a wide variety of inlet conditions and to calculate extinction limits. The surrogate model includes a semi-detailed kinetic mechanism composed of 221 gaseous species participating in 5032 reactions. Experimentally, counterflow diffusion flames are established, in which multicomponent fuel vaporization is achieved through the use of an ultrasonic nebulizer that introduces small fuel droplets into a heated nitrogen stream, fostering complete vaporization without fractional distillation. Temperature profiles and extinction limits are measured in all flames and compared with predictions using the semi-detailed mechanism. These measurements show good agreement with predictions in single-component n-dodecane, methylcyclohexane, and iso-octane flames. Good agreement also exists between predicted and measured variables in flames of the surrogate, and the agreement is even better between the experimental JP-8 flames and the surrogate predictions.
- A Multi-scale Computational Approach for Nanoparticle Growth in Combustion EnvironmentsAngela Violi and Gregory A. VothIn High Performance Computing and Communications, Jan 2005
In this paper a new and powerful computer simulation capability for the characterization of carbonaceous nanoparticle assemblies across multiple, connected scales, starting from the molecular scale is presented. The goal is to provide a computational infrastructure that can reveal through multi-scale computer simulation how chemistry can influence the structure and function of carbonaceous assemblies at significantly larger length and time scales. Atomistic simulation methodologies, such as Molecular Dynamics and Kinetic Monte Carlo, are used to describe the particle growth and the different spatial and temporal scales are connected in a multi-scale fashion so that key information is passed upward in scale. The modeling of the multiple scales are allowed to be dynamically coupled within a single computer simulation using the latest generation MPI protocol within a grid-based computing scheme.
- The relative roles of acetylene and aromatic precursors during soot particle inceptionAngela Violi, Gregory A. Voth, and Adel F. SarofimProceedings of the Combustion Institute, Jan 2005
Kinetic Monte Carlo (KMC) and Molecular Dynamic (MD) methodologies were applied sequentially to simulate soot inception in premixed fuel-rich benzene flames to identify the role of accessibility to reaction sites on the relative rate of reactions of molecules of different size. This approach is designed to provide atomistic scale structure (bonds, bond-angles, and dihedral angles) as soot precursors evolve into a three-dimensional structure. Two different times in a benzene flame, experimentally studied by Homann were chosen to examine the reaction sequence between 20 gas phase species and the soot seed, starting with pyrene. The curvature of the seed particle is found to increase with particle size becoming significant when the number of carbon atoms exceed 100 and with the seed developing a spheroidal shape when the number of carbon atoms exceeds 400. The curvature increases with temperature in the flame, being greater at 2100 than at 1700K. The accessibility of the sites on the growing seed molecule decreases with increases in curvature of the seed particle and size of the reacting molecule. The accessibility decreases from 1 at a seed size of around 100–150 carbons atoms down to about 0.5 at a size of 400 carbon atoms for acetylene and down to about 0.3 at a seed size of 200 atoms for acenaphthylene. These results show the value of the KMC/MD methodology, which provides insights into the details of soot particle inception.
- The relative roles of acetylene and aromatic precursors during soot particle inceptionAngela Violi, Gregory A. Voth, and Adel F. SarofimProceedings of the Combustion Institute, Jan 2005
Kinetic Monte Carlo (KMC) and Molecular Dynamic (MD) methodologies were applied sequentially to simulate soot inception in premixed fuel-rich benzene flames to identify the role of accessibility to reaction sites on the relative rate of reactions of molecules of different size. This approach is designed to provide atomistic scale structure (bonds, bond-angles, and dihedral angles) as soot precursors evolve into a three-dimensional structure. Two different times in a benzene flame, experimentally studied by Homann were chosen to examine the reaction sequence between 20 gas phase species and the soot seed, starting with pyrene. The curvature of the seed particle is found to increase with particle size becoming significant when the number of carbon atoms exceed 100 and with the seed developing a spheroidal shape when the number of carbon atoms exceeds 400. The curvature increases with temperature in the flame, being greater at 2100 than at 1700K. The accessibility of the sites on the growing seed molecule decreases with increases in curvature of the seed particle and size of the reacting molecule. The accessibility decreases from 1 at a seed size of around 100–150 carbons atoms down to about 0.5 at a size of 400 carbon atoms for acetylene and down to about 0.3 at a seed size of 200 atoms for acenaphthylene. These results show the value of the KMC/MD methodology, which provides insights into the details of soot particle inception.
2004
- Kinetics of Hydrogen Abstraction Reactions from Polycyclic Aromatic Hydrocarbons by H AtomsAngela Violi, Thanh N. Truong, and Adel F. SarofimThe Journal of Physical Chemistry A, Jun 2004
An application of the Reaction Class Transition State Theory/Linear Energy Relationship (RC-TST/LER) is presented for the evaluation of the thermal rate constants of hydrogen abstraction reactions by H atoms from Polycyclic Aromatic Hydrocarbons (PAH). Two classes of reactions have been considered, namely hydrogen bonded to six- and five-membered rings, respectively, and twenty-two reactions have been used to develop the RC-TST/LER parameters. B3LYP and BH&HLYP density functional theory methods were used to calculate necessary potential energy surface information. Detailed analyses of RC-TST/LER reaction factors lead to the conclusion that rate constants for any reaction in these two classes can be approximated by those of its corresponding principal reaction corrected by the reaction symmetry factor. Specifically, for hydrogen abstraction from six-membered rings such as naphthalene and pyrene, k(T) = (σ/\σ\H+C6H6)\thinspacekH+C6H6 = (\σ\/6){1.42 \texttimes 108T1.77 exp(-6570/T)}(cm3/mol\textperiodcentereds), and for hydrogen abstraction from five-membered rings such as acenaphthylene and acephenanthrylene, k(T) = (\σ\/\σ\H+C12H8)\thinspacekH+C12H8 = (\σ\/2){3.27 \texttimes 108T1.71 exp(-8170/T)} (cm3/mol\textperiodcentereds), where \σis the reaction symmetry number.
- Kinetic Monte Carlo–Molecular Dynamics Approach To Model Soot InceptionAdel F. Sarofim Angela Violi and Gregory A. VothCombustion Science and Technology, Jun 2004
The processes involved in soot precursor formation exhibit a wide range of timescales, spanning pico- or nanoseconds for intramolecular processes that can occur on a particle surface to milliseconds for the formation of the first soot precursors. To accurately describe the soot formation process, it is important to model the reactions happening at different timescales. The use of atomistic models allows this. The code, named KMC/MD, combines the strengths of Kinetic Monte Carlo for long-time sampling, and Molecular Dynamics for relaxation processes. It enables the investigation of physical as well as chemical properties of the carbonaceous nanoparticles formed, such as particle morphology and concentration of free radicals.
- A Kinetic Model Of Particulate Carbon Formation In Rich Premixed Flames Of Ethylene And BenzeneAngela Violi, A D’Anna, and A D’AlessioInternational Journal of Energy for a Clean Environment, Jun 2004
A detailed chemical kinetic mechanism, which emphasizes the role of resonantly stabilized radicals in the growth of aromatic species, has been used to study the effect of fuel type on the formation of total particulate carbon in rich premixed flames. The model has been able to predict the concentration and formation rate of total organic carbon collected in sooting rich flames of ethylene and benzene. Model results have shown that the combination of resonantly stabilized radicals is the controlling step in the formation of aromatics in flames; in particular, cyclopentadienyl radical self-combination is the dominant route of multi-ring aromatic formation. The larger amount of cyclopentadienyl radicals in benzene fuel oxidation may explain the larger yield of particulate carbon formed in aromatic fuel flames than aliphatic ones in the same conditions of pressures, temperatures, and flux of carbon in surplus with respect to the stoichiometric one.
- Modeling of soot particle inception in aromatic and aliphatic premixed flamesAngela VioliCombustion and Flame, Jun 2004
The growth of hydrocarbon molecules up to sizes of incipient soot is computed in premixed laminar flames using kinetic Monte Carlo and molecular dynamic methodologies (AMPI code). This approach is designed to preserve atomistic scale structure (bonds, bond angles, dihedral angles) as soot precursors evolve into three-dimensional structures. Application of this code to aliphatic (acetylene) and aromatic (benzene) flame environments is able to explain results in the literature on the differences in properties of soot precursors from these two classes of flames, particularly relating to H/C ratio, particle sphericity, and depolarization ratio.
2003
- Large eddy simulations of accidental fires using massively parallel computersPhilip Smith, Rajesh Rawat, Jennifer Spinti, Seshadri Kumar, Stanislav Borodai, and 1 more authorIn 16th AIAA Computational Fluid Dynamics Conference, Jun 2003
Large-scale hydrocarbon pool-fires (liquid hydrocarbon fires of pool diameters > 10m) are difficult to analyze experimentally because of the scale of the events. Massively parallel computing using thousands of processors offers a vehicle to deliver simulation-science based analysis of such large-scale events. On these scales the fires are turbulent; that is, dynamic vortical structures are present. The fire chemistry requires reaction mechanisms that allow for soot formation, growth and oxidation. Radiation, the dominant mode of heat transfer, is strongly affected by the presence of soot. Large Eddy Simulation (LES) provide temporally and spatially resolved information over time scales that include the well known puffing frequency and overall burning time for these fires and over spatial scales that include the vortical structures on the order of 10cm in scale and larger. Smaller spatial and temporal scale (subgrid-scale) processes are modeled. Verification and validation of the LES fire simulation tool with its subgrid-scale models provides some confidence that simulations replicate observables. Under funding by the United States Department of Energy Advanced Simulation and Computing Program (ASCI), we have used a 20m heptane pool fire as a prototype target for demonstrating the use of high performance computers to integrate multi-scale phenomena. This simulation tool with its appropriate subgrid-scale models has been built within the Uintah Computational Framework (UCF), a component-based Problem Solving Environment (PSE) that provides the framework for large-scale parallelization. Parallelization scalability studies of the simulation tool reveal close to linear scalability up to 1000 processors. Kinetic Monte Carlo methods have been combined with Molecular Dynamics (KMC/MD) to compute chemical structural growth of soot. Simulations that combine these ranges of scales have reproduced features of large-scale fires such as initial roll-up of vortices and subsequent breakup from the continuous flame zone. The sensitivity of the radiant heat transfer to the soot formation mechanism in these large scale fires has been studied. \textcopyright 2003 by Philip John Smith.
- Modeling aerosol formation in opposed-flow diffusion flamesAngela Violi, Andrea D’Anna, Antonio D’Alessio, and Adel F SarofimChemosphere, Jun 2003Pic7
The microstructures of atmospheric pressure, counter-flow, sooting, flat, laminar ethylene diffusion flames have been studied numerically by using a new kinetic model developed for hydrocarbon oxidation and pyrolysis. Modeling results are in reasonable agreement with experimental data in terms of concentration profiles of stable species and gas-phase aromatic compounds. Modeling results are used to analyze the controlling steps of aromatic formation and soot growth in counter-flow configurations. The formation of high molecular mass aromatics in diffusion controlled conditions is restricted to a narrow area close to the flame front where these species reach a molecular weight of about 1000 u. Depending on the flame configuration, soot formation is controlled by the coagulation of nanoparticles or by the addition of PAH to soot nuclei.
- A time-scale problem for the formation of soot precursors in premixed flamesAngela Violi, GA Voth, and AF SarofimIn , Jun 2003
A Kinetic Monte Carlo (KMC) and Molecular Dynamics (MD) study on soot inception in aliphatic and aromatic flames was carried out. Acetylene, hydrogen, naphthalene, and acenaphthylene species were considered in the study as they are key contributors to the PAH growth process and used as input to the KMC/MD code. Formation of carbonaceous nanoparticles could be followed in a chemically specific way. Properties of the particles, such as H/C ratio and morphology, were computed, showing the presence of elongated shapes for young nanoparticles. The influence of two different environments, an aromatic and aliphatic flame, was assessed in terms of structures of the particles formed. The use of this approach would allow the investigation on the physical, e.g., porosity, density, sphericity, as well as chemical, e.g., H/C, aromatic moieties, number of cross-links, properties.
2002
- Experimental formulation and kinetic model for JP-8 surrogate mixturesCombustion Science and Technology, Jun 2002
Jet A and JP-8 are kerosene fuels used in aviation and consist of complex mixtures of higher order hydrocarbons, including alkanes, cycloalkanes, and aromatic molecules. The objectives of the current work are to develop a surrogate mixture to represent JP-8 fuels and to discuss a general detailed chemical kinetic model for jet fuels, which is suitable for future reduction. Asurrogate blend of six pure hydrocarbons is found to adequately simulate the distillation and compositional characteristics of a practical JP-8. A hierarchically constructed kinetic model already available for the oxidation of alkanes and simple aromatic molecules (benzene, toluene, ethylbenzene, xylene, etc.) is extended to include methylcyclohexane and tetralin as new reference fuel components. The kinetic model is validated through comparisons with experimental data for the pure components and it is also used to verify and predict the structures of laminar premixed flames of different pure components as well as conventional kerosene fuels.
- Mechanistic pathways to explain H/C ratio of soot precursorsA. F. Sarofim A. Violi and T. N. TruongCombustion Science and Technology, Jun 2002
Pathways for the growth of high-molecular-mass compounds are presented, showing how reactions between aromatic moieties can explain recent experimental findings. A fundamental molecular analysis of polycyclic aromatic hydrocarbon growth processes in combustion systems involving five-membered ring compounds is presented using quantum mechanical density functional methods. Higher aromatics are produced through a two-step radical-molecule addition reaction and the iteration of this mechanism followed by rearrangement of the carbon framework ultimately leads to high-molecular-mass compounds. The distinguishing features of the proposed model lie in the chemical specificity of the routes considered. Naphthalene and acenaphthylene are used as examples of the aromatic and cyclopentafused aromatic classes of compounds postulated to be of importance in molecular weight growth. These reaction pathways are analyzed with a view toward explaining recent experimental findings on H/C ratio, NMR, and LMMS of soot precursors.
- A fully integrated kinetic monte carlo/molecular dynamics approach for the simulation of soot precursor growthA. Violi, A. Kubota, T.N. Truong, W.J. Pitz, C.K. Westbrook, and 1 more authorProceedings of the Combustion Institute, Jun 2002
The emphasis in this paper is on presenting a new methodology, together with some illustrative applications, for the study of polycyclic aromatic hydrocarbon polymerization leading to soot, widely recognized as a very important and challenging combustion problem. The new code, named fully integrated Kinetic Monte Carlo/Molecular Dynamics (KMC/MD), places the two simulation procedures on an equal footing and involves alternating between KMC and MD steps during the simulation. The KMC/MD simulations are used in conjunction with high-level quantum chemical calculations. With traditional kinetic rates and dealing with the growth of particles, it is often necessary to perform a lurnping procedure in which much atomic-scale information is lost. Our KMC/MD approach is designed to preserve atomic-scale structure: a single particle evolves in time with real three-dimensional structure (bonds, bond angles, dihedralangles). In this paper, the methodology is illustrated by a sample simulation of high molecular mass compound growth in an environment (T, H, H2, naphthalene, and acenaphthylene concentrations) of a low-pressure laminar premixed benzene/oxygen/argon flame with an equivalence ratio of 1.8. The use of this approach enables the investigation of the physical (e.g., porosity, density, sphericity) as well as chemical (e.g. H/C, aromatic moieties, number of cross-links) properties.
- Fully-integrated molecular dynamics kinetic Monte Carlo code: A new tool for the study of soot precursor growth in combustion conditionsAngela Violi, A Kubota, W Pitz, CK Westbrook, and AF SarofimIn , Jun 2002
A new tool, which combines two common methods, i.e., Kinetic Monte Carlo (KMC) and Molecular Dynamics (MD), for examining the molecular transformations from gas phase precursors to soot-like material was introduced. In the new code, named Fully-Integrated KMC/MD, puts the two simulation procedures (KMC and MD) on an equal footing and involves alternating between MD and KMC steps during the simulation. The code was used to analyze some experimental results obtained by Prof. Homann’s research group. The environment chosen for this analysis is a low-pressure laminar premixed benzene/oxygen/argon flame. The open structures obtained with the reaction scheme and the code provide an explanation for the H/C ratios of soot precursors and young soots being greatly higher than those of polybenzenoid structures of equal molecular weight. The curvature introduced by forming five- and six-membered rings is the most noticeable feature of these structures.
2001
- A reaction pathway for nanoparticle formation in rich premixed flamesA D’Anna, A Violi, A D’Alessio, and A.F SarofimCombustion and Flame, Jun 2001
Aromatics growth beyond 2-, 3-ring PAH is analyzed through a radical-molecule reaction mechanism which, in combination with a previously developed PAH model, is able to predict the size distribution of aromatic structures formed in rich premixed flames of ethylene at atmospheric pressure with C/O ratios across the soot threshold limit. Modeling results are in good agreement with experimental data and are used to interpret the ultraviolet absorption and the light scattering measured in flames before soot inception. The model shows that the total number concentration of high molecular mass aromatics and the different moments of the size distribution are functions of both the PAH and H-atom concentrations, two quantities which have different trends as functions of the residence time and the C/O ratio. Regimes of nearly stoichiometric or slightly rich premixed combustion are dominated by reactions between aromatics which lead to the formation of particles with sizes of the order of 3 to 4 nm. At higher C/O ratios the formation of nanoparticles is less efficient. Particles with sizes of the order of 2 nm are predicted in flames at the threshold of soot formation, whereas particles with sizes around 1 to 1.5 nm are predicted in fully sooting conditions.
- A modeling evaluation of the effect of chlorine on the formation of particulate matter in combustionAngela Violi, Andrea D’Anna, and Antonio D’AlessioChemosphere, Jun 2001Proceedings of the 6th Intl Congress on Toxic combustion
The effect of chlorine on the fuel-rich oxidation of hydrocarbons and on the molecular weight growth of aromatics is analyzed by simulating experiments featuring a model chlorinated additive CH3Cl in a jet-stirred/plug-flow reactor and premixed flames. The kinetic model used in this work emphasizes the role of resonantly stabilized radicals in the formation and growth of aromatics, and considers soot inception as the net effect of molecular weight growth and graphitization of aromatic structures. Chlorinated hydrocarbons decompose at temperatures significantly lower than hydrocarbons, producing reactive Cl-atoms, which have a strong tendency to go to HCl. The HCl, tying up the H-atoms, inhibits hydrocarbon oxidation. The model is able to predict not only the levels but the shape of the experiments quite well and also the surprising finding of an increased soot formation associated with lower PAH levels found in rich flames with significant levels of chlorine. Based on reaction kinetic analysis, chlorine addition to the fuel enhances soot formation by promoting the formation of aromatic-ring compounds and accelerating the abstraction of aromatic H-atoms from stable PAH molecules. This process activates the transformation of aromatics to soot.
- Modelling of particulate formation in opposed diffusion flamesAngela Violi, AF Sarofim, A D’Anna, and A D’AlessioIn , Jun 2001
The microstructures of atmospheric pressure, counter-flow, sooting, flat, laminar ethylene diffusion flames have been studied numerically. Aromatics growth beyond 2-, 3-ring PAH is analyzed through a radical-molecule reaction mechanism which, in combination with a previously developed PAH model, is able to predict the concentration profiles of aromatic structures formed in flames. Modelling results are in good agreement with experimental data and are used to interpret the soot volume fraction and PAH fluorescence measured in flames.
- Quantum mechanical study of molecular weight growth process by combination of aromatic moleculesA Violi, A.F Sarofim, and T.N TruongCombustion and Flame, Jun 2001
Formation pathways for high-molecular-mass compound growth are presented, showing why reactions between aromatic moieties are needed to explain recent experimental findings. These reactions are then analyzed by using quantum mechanical density functional methods. A sequence of chemical reactions between aromatic compounds (e.g., phenyl) and compounds containing conjugated double bonds (e.g., acenaphthylene) was studied in detail. The sequence begins with the H-abstraction from acenaphthylene to produce the corresponding radical, which then furnishes higher aromatics through either a two-step radical-molecule reaction or a direct radical-radical addition to another aromatic radical. Iteration of this mechanism followed by rearrangement of the carbon framework ultimately leads to high-molecular-mass compounds. This sequence can be repeated for the formation of high-molecular-mass compounds. The distinguishing features of the proposed model lie in the chemical specificity of the routes considered. The aromatic radical attacks the double bond of five-membered-ring polycyclic aromatic hydrocarbons. This involves specific compounds that are exceptional soot precursors as they form resonantly stabilized radical intermediates, relieving part of the large strain in the five-membered rings by formation of linear aggregates.
- The Validation Web Site: A Combustion Collaboratory over the InternetAngela Violi, Xiaodong Chen, Gary Lindstrom, Eric Eddings, and Adel F. SarofimIn Computational Science - ICCS 2001, Jun 2001
The Soot Model Development Web Site (SMDWS) is a project to develop collaborative technologies serving combustion researchers in DOE national laboratories, academia and research institutions throughout the world. The result is a shared forum for problem exploration in combustion research. Researchers collaborate over the Internet using SMDWS tools, which include a server for executing combustion models, web-accessible data storage for sharing experimental and modeling data, and graphical visualization of combustion effects. In this paper the authors describe the current status of the SMDWS project, as well as continuing goals for enhanced functionality, modes of collaboration, and community building.
2000
- On the relevance of surface growth in soot formation in premixed flamesA. D’Alessio, A. D’Anna, P. Minutolo, L.A. Sgro, and A. VioliProceedings of the Combustion Institute, Jun 2000
The role of surface growth mechanisms in particle mass accumulation was investigated in rich, premixed, ethylene/air flames from non-sooting to moderately sooting conditions using in situ optical diagnostics and predictions from a detailed chemical kinetic model. Particles formed just after the flame front, which are transparent to the visible light but absorb in the UV range, have been detected in large amounts in non-sooting flames and earlier in the flame than soot particles in sooting-flames, using UV-visible light optical diagnostics. For C/O<0.8, the amount of visible-transparent particles accounts for the total mass of soot detected later in the flame, indicating that surface growth processes are negligible and that soot formation is a rearrangement of the carbonaceous material already present in the form of smaller particles. Furthermore, predictions from the kinetic model, which does not include surface growth reactions, agree well with experiments for C/O<0.8. The model is able to predict the total carbon contained in particles observed in the non-sooting flames and in slightly sooting flames, as well as that observed up to the onset of soot formation in richer flames. For C/O>0.8, additional soot growth mechanisms need to be included in the mechanism to account for the amount of soot observed in the later part of the flames. Interestingly enough, this late growth mechanism occurs almost simultaneously with a strong increase in the coagulation rate of the particles, thus indicating that both effects are related and are probably due to a major change in the chemical nature of the particle surface.
- Modeling the rich combustion of aliphatic hydrocarbonsA D’Anna, Angela Violi, and A D’AlessioJun 2000
A new kinetic mechanism has been developed for the formation of benzene and high-molecular-mass aromatic compounds in rich flames of aliphatic hydrocarbons. The kinetic scheme emphasizes both the role of resonantly stabilized radicals in the growth of aromatics and the standard acetylene addition mechanism. The model has been used to simulate premixed flames of acetylene and ethylene where the concentrations of radicals and high-molecular-mass compounds are known. The kinetic scheme accurately reproduces the concentrations and trends of radicals and stable species including benzene and total aromatic compounds. Formation of benzene is controlled by propargyl radical combination. The model reproduces well the profiles of benzene for the different hydrocarbons and in the different operating conditions. Key reactions in the formation of high-molecular-mass aromatics are the combinations of resonantly stabilized radicals, including cyclopentadienyl self-combination, propargyl addition to benzyl radicals, and the sequential addition of propargyl radicals to aromatic rings. The predicted amounts of total aromatic compounds increase at the flame front and remain constant in the postoxidation zone of the flames, attaining the final concentrations of soot, in slightly-sooting conditions. As a consequence, the carbonaceous species which contribute to soot formation are already present at the flame front as high-molecular-mass structures. Soot is formed through dehydrogenation and aromatization of the high-molecular-mass compounds, rather than by surface growth. Copyright (C) 2000 The Combustion Institute.
- Experimental and modeling study of particulate formation in high-pressure diesel-like conditionsAngela Violi, Andrea D’Anna, Antonio D’Alessio, Massimo Astarita, and Bianca Maria VagliecoProceedings of the Combustion Institute, Jun 2000
The diesel combustion process has been characterized by combined measurements of flame intensity and extinction in the UV-visible range in an optically accessible divided-chamber diesel engine at different air/fuel ratios and constant engine speed, using n-heptane and commercial diesel oil as fuels. The analysis of the diesel combustion performed by multiwavelength extinction measurements has shown that in the high-pressure conditions typical of diesel combustion, two classes of compounds are formed: soot particles and organic aerosol. The latter structures are present just after fuel ignition, and their concentration suddenly increases as the combustion proceeds, reaching a maximum value well before the formation of soot particles, which occurs after an induction period of about 1 ms. A detailed kinetic mechanism previously developed to predict the total concentration of particulate carbon in rich flames of aliphatic hydrocarbons has been used to model the formation of particulates in diesel combustion. The kinetic scheme, coupled to a simplified model of diesel combustion, has been able to correctly predict the total concentration and the initial fast formation rate of aromatics.
1999
- Modeling of particulate formation in combustion and pyrolysisAngela Violi, Andrea D’Anna, and Antonio D’AlessioChemical Engineering Science, Jun 1999
We use a detailed chemical kinetic mechanism to explore the effects of C/O ratio, temperature and pressure on the formation of high molecular weight aromatic species in premixed flames and shock tubes in a wide range of operating conditions. Key sequences of reactions in the formation of aromatics are the addition of acetylene to smaller aromatics, activated by H-atom abstraction and the combination of resonantly stabilized radicals, including cyclopentadienyl radical combination, propargyl addition to benzyl radicals and the sequential addition of propargyl radicals to aromatic rings. The modeling results are compared with those obtained by considering only the H-abstraction acetylene addition (HACA) route for aromatic formation to identify the controlling steps in different combustion regimes and to assess the viability of the HACA and resonantly stabilized radical (RSR) pathways to model aromatic growth. The full model is able to predict the concentration and formation rate of total organic carbon collected in slightly sooting rich flames at different temperatures and pressures. On the contrary, the HACA model predicts concentrations and formation rates more than one order of magnitude lower than those measured in experiments. This result indicates that a combination of resonantly stabilized radicals are the controlling steps in the formation of aromatics in flames. The oxidative environment is pivotal in the formation of aromatics to activate the pathways involving the resonantly stabilized radicals in the aromatic growth process. Since the model simulates only the formation of two- and three-ring aromatics, it can be hypothesized that total organic material collected in flames, i.e. a mass quantity much larger than the chromatographable polycyclic aromatic hydrocarbons, is the result of a fast reactive coagulation of small aromatics, forming structures of high molecular mass. Results indicate that reactions leading to the formation of two- and three-ring aromatics are rate-limiting, and combination reactions involving these aromatics control soot formation in oxidative or slightly sooting regimes. In very fuel-rich and pure pyrolysis conditions, the two models predict the same amount and formation rate of aromatics; this indicates that the HACA route controls formation in these conditions since the RSR pathways are activated only in oxidative environments.
1998
- A kinetic model for the formation of aromatic hydrocarbons in premixed laminar flamesAndrea D’Anna and Angela VioliSymposium (International) on Combustion, Jun 1998Twenty-Seventh Sysposium (International) on Combustion Volume One
A detailed chemical kinetic mechanism, including 340 elementary steps and 90 species, has been developed to simulate the formation of aromatic compounds in rich premixed flames of aliphatic hydrocarbons. The mechanism can reproduce the concentration profiles and net rates of benzene and larger aromatic hydrocarbons (two- and three-ring polycyclic aromatic hydrocarbons [PAHs]) in a wide range of temperatures for a slightly sooting, premixed ethylene-oxygen flames. Key sequences of reactions in the formation of aromatics are the combination of resonantly stabilized radicals, whereas the alternative mechanism that involves acetylene addition does not seem to be fast enough to explain the observed formation rates of aromatics in the flames examined. The main routes involved in the formation of the first aromatic ring are the propargyl self-combination and its addition to 1-methylallenyl radicals. Cyclopentadienyl radical combination, propargyl addition to benzyl radicals, and the sequential addition of propargyl radicals to aromatic rings are the controlling steps for the formation of larger aromatic species. The predicted concentration and formation rate of larger aromatics are higher than those of PAHs identified by gas chromatography but similar to those of total aromatic species collected in flames, namely, soot and tar-like material. This result suggests that tar-like material can be considered as the result of a fast reactive coagulation of two- and three-ring PAHs, which form structures with aromatic compounds, eventually connected by aliphatic bonding. Soot inception may be the result of an internal rearrangement of tar without significant contribution of light hydrocarbons, at least in the conditions examined. The model has been used to explore the effect of C/O ratio and temperature on the formation rate and concentration of aromatic hydrocarbons. The net formation rate of aromatics increases monotonically with C/O ratio, and at a fixed C/O ratio, it passes through a maximum as temperature is increased. A comparison with the “bell-shaped” concentration profile of soot collected in flames at different temperatures supports the hypothesis that the formation of two- and three-ring structures may be the rate-determining step in soot formation for slightly sooting regimes.
1996
- The effect of temperature on soot inception in premixed ethylene flamesA Ciajolo, A D’anna, R Barbella, A Tregrossi, and Angela VioliIn , Jun 1996
The temperature dependence of soot and related hydrocarbon formation was studied in fuel-rich, premixed C2H4/O2 flames by changing the flame temperature but keeping a constant C/O ratio. Three flames with temperature levels between the low-and high-temperature soot threshold of the bell-shaped domain of soot formation were analyzed. In addition to the formation of pyrolytic species such as acetylene, benzene, polycyclin aromatic compounds (PAHs), and soot, the large formation of a tarlike material was revealed in the soot preinception region of the flames. The chemical analysis and spectroscopic characterization of these species showed that tar is made up of high-molecular-weight compounds in which aliphatic and aromatic functionalities coexist. The decrease in the concentration and the aromatic character of tar was found to correspond with soot inception, indicating that it could be responsible for soot nucleation. PAH molecules indirectly participate in soot inception, providing the building blocks of tar and/or participating in further soot surface growth. The influence of temperature on soot formation was found to be different at the extremes of the bell-shaped domain of soot. In the flame near the low-temperature soot threshold, benzene, PAHs and pyrolytic species are formed in larger amounts, but low temperatures hinder the reactive coagulation of PAH molecules and aliphatic radicals into tarlike material, hindering, in turn, soot production. The buildup of tarlike material and the process of aromatization occur too slowly to allow the massive soot inception if the flame temperature is not high enough. At higher temperatures, oxidation is very active in the secondary oxidation zone of the flame, thus destroying PAHs that are not then available for formation of tarlike species. Consequently, soot inception is reduced. In the intermediate-temperature flame, where maximum soot formation was found, the temperature level is ideal for the formation of both PAHs and tar and their transformation to soot particles. \textcopyright 1996 Combustion Institute.